Synopsis of Research Program
THE REDOX REGULATION OF CANCER CELL FATE
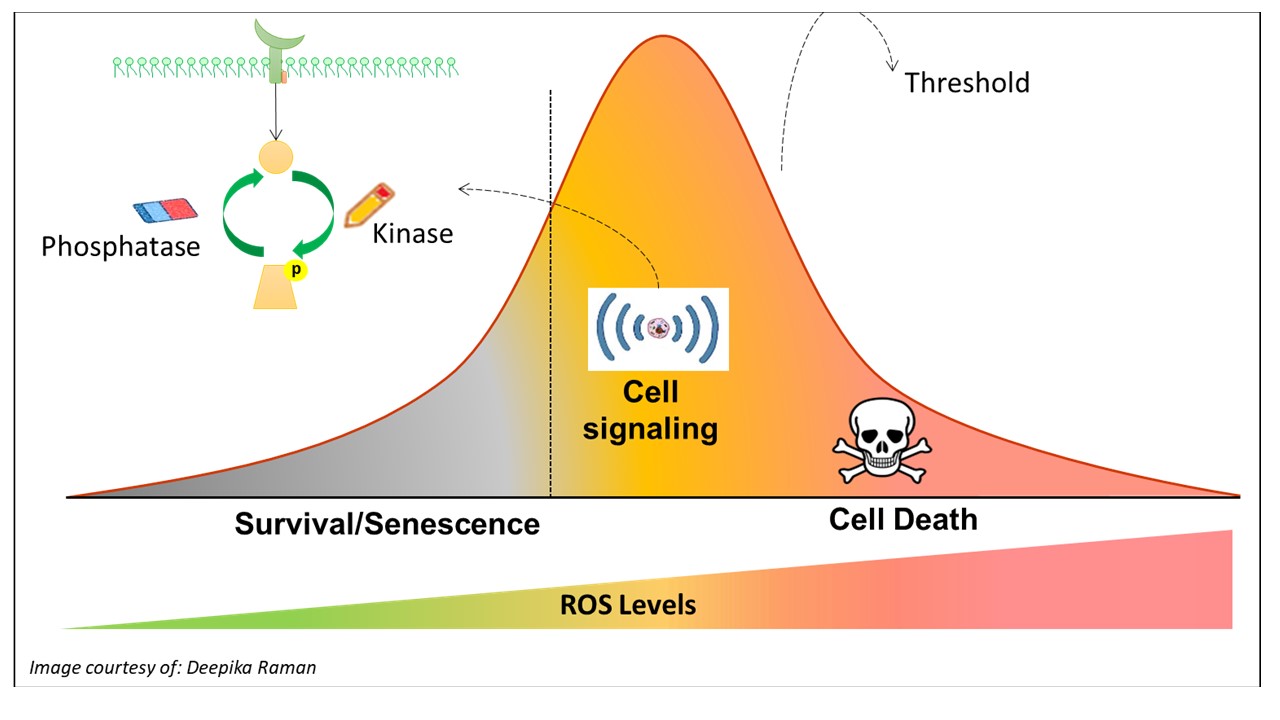
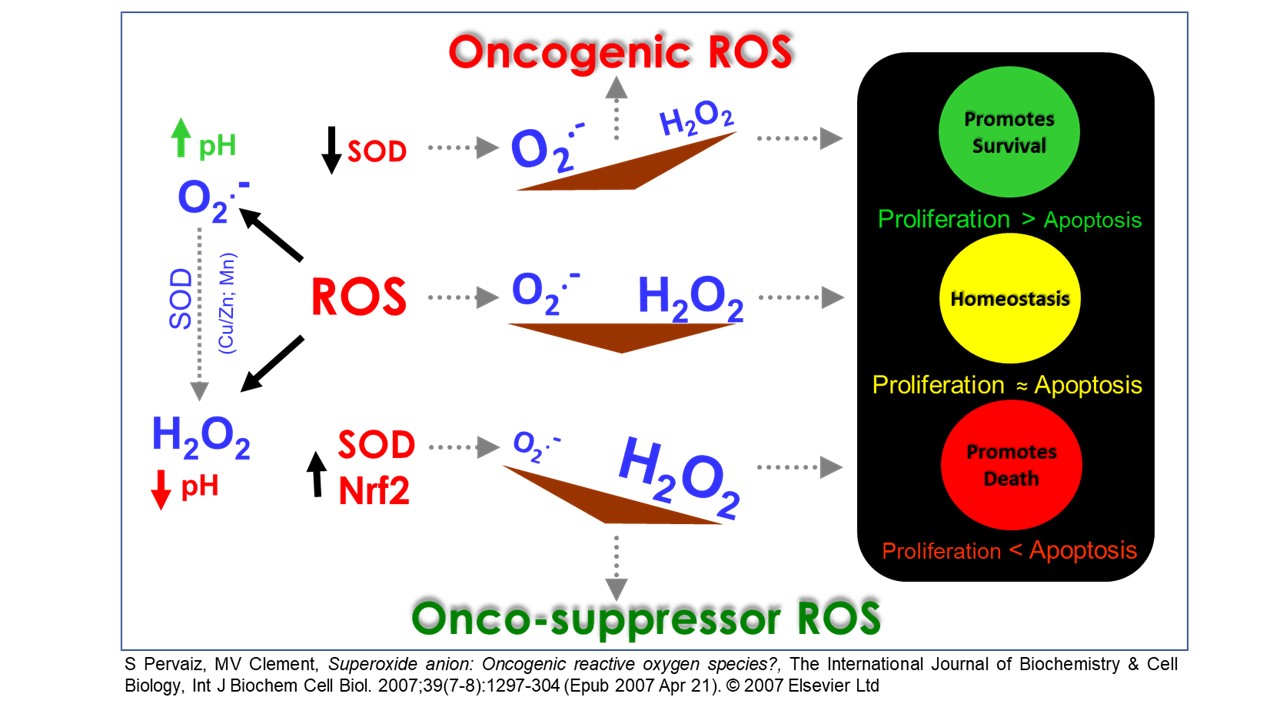
The increase in intracellular reactive oxygen species (ROS) has been shown to promote cell survival, proliferation or senescence via the activation of various kinases and/or the inactivation of phosphatases. However, an increase that overwhelms the antioxidant defence system may in turn inflict oxidative stress, biomolecular damage or even cell death. Over the years, the focus of our research has been to unravel or identify the specific pathways that govern the different redox spectrums of cell death and survival, particularly from the standpoint of tumorigenesis.
On this end, our work has demonstrated that a non-toxic increase in intracellular ROS level, in favour of the superoxide anion (O2-), is capable of promoting survival in the presence of death-inducing stimuli. On the other hand, an increase of ROS that favors the dismutated product of O2.- - hydrogen peroxide (H2O2) could promote apoptotic cell death. These findings have thus paved the direction of our research in further unravelling the different redox pathways that regulate cancer cell fate.
A: THE PRO-SURVIVAL FUNCTION OF ROS
A1. Novel functional biology of the anti-apoptotic protein Bcl-2
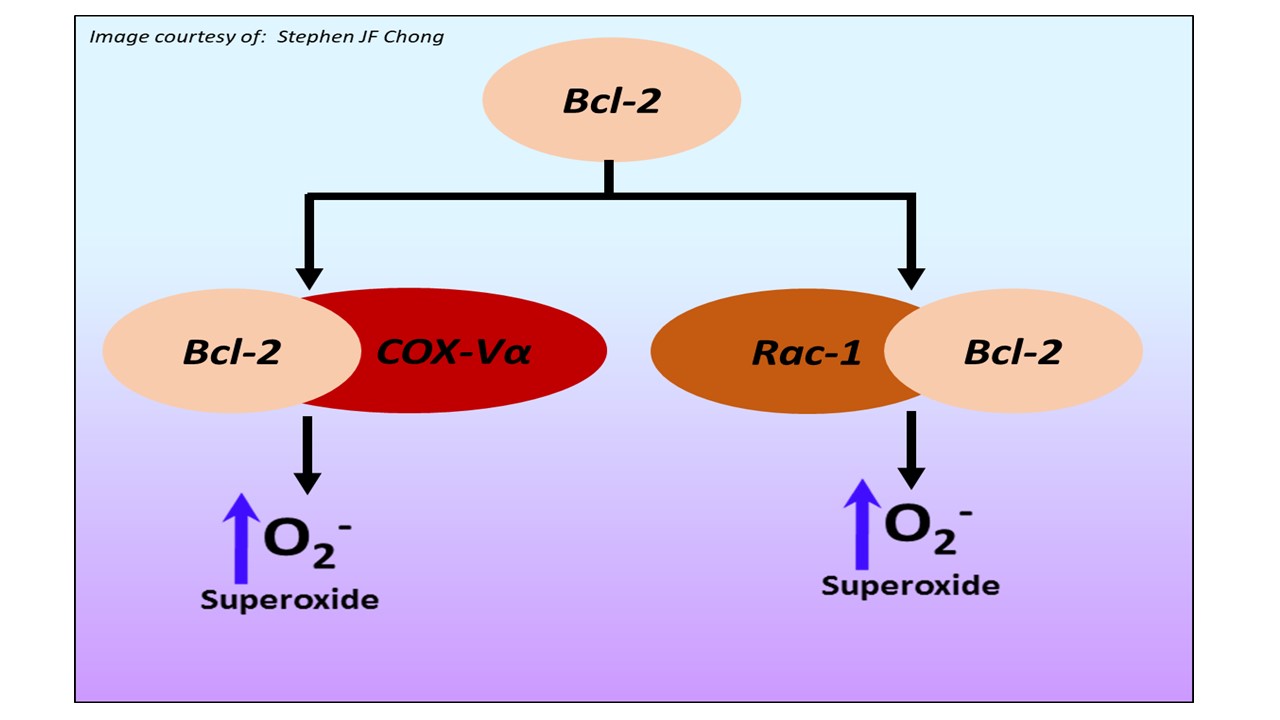
The Bcl-2 family of proteins regulates apoptotic signaling in cancer cells. The anti-apoptotic members such as Bcl-2 and Bcl-xL inhibit mitochondrial death amplification signals, thereby rendering cancer cells refractory to chemotherapy. Our work over the years has highlighted a novel facet in the biology of Bcl-2 by linking its anti-apoptotic activity to a change in cellular redox milieu. Bcl-2-induced resistance to apoptosis was associated with an increase in intracellular superoxide anion, and more importantly, pharmacological or genetic approaches to clamp down intracellular superoxide reverted cell sensitivity to apoptosis stimuli without compromising the mitochondrial stabilizing activity of Bcl-2 (Cell Death Diff. 2003). Subsequent efforts to decipher the underlying mechanisms of the pro-oxidant activity of Bcl-2 revealed evidence implicating increases in mitochondrial oxygen consumption and cytochrome C oxidase (complex IV of electron transport chain) activity in a variety of human cell lines overexpressing Bcl-2 (Cell Death Diff. 2007). Cytochrome C oxidase is the rate-limiting enzyme in the mitochondrial electron transport chain and comprises of 13 sub-units, 10 of which are nuclear transcribed and imported into the mitochondria. Interestingly, the significant increase in the activity of Cytochrome C oxidase upon Bcl-2 overexpression is a function of a novel physical interaction between Bcl-2 and the Vα sub-unit of Cytochrome C oxidase and its enhanced import into the mitochondria during normoxic states (Cell Death Diff. 2010). Our continued efforts to unravel the non-canonical activity of Bcl-2 revealed the small GTPase Rac1 as a novel binding partner of Bcl-2. Not only did we identify the putative binding domain within Bcl-2 responsible for this interaction, but also provided evidence that interrupting this interaction alleviated the effects of Bcl-2 on mitochondrial superoxide production and drug-induced apoptosis. Of note, an increase in Bcl-2-Rac1 interaction was demonstrated in clinical samples from B cell lymphomas, thus providing translational relevance to these findings (Blood, 2011). We have also identified the non-structured loop region within Bcl-2 (contains important phosphorylation sites) as critical in its interaction with GTP-bound active Rac1, and interestingly provided evidence that STAT-3 was a critical downstream mediator of Bcl-2-Rac1 interaction-induced survival response (Oncotarget, 2015). On the backdrop of these findings, our ongoing studies and future investigations are focused on identifying downstream targets of redox-mediated cell survival signaling with potential therapeutic relevance.
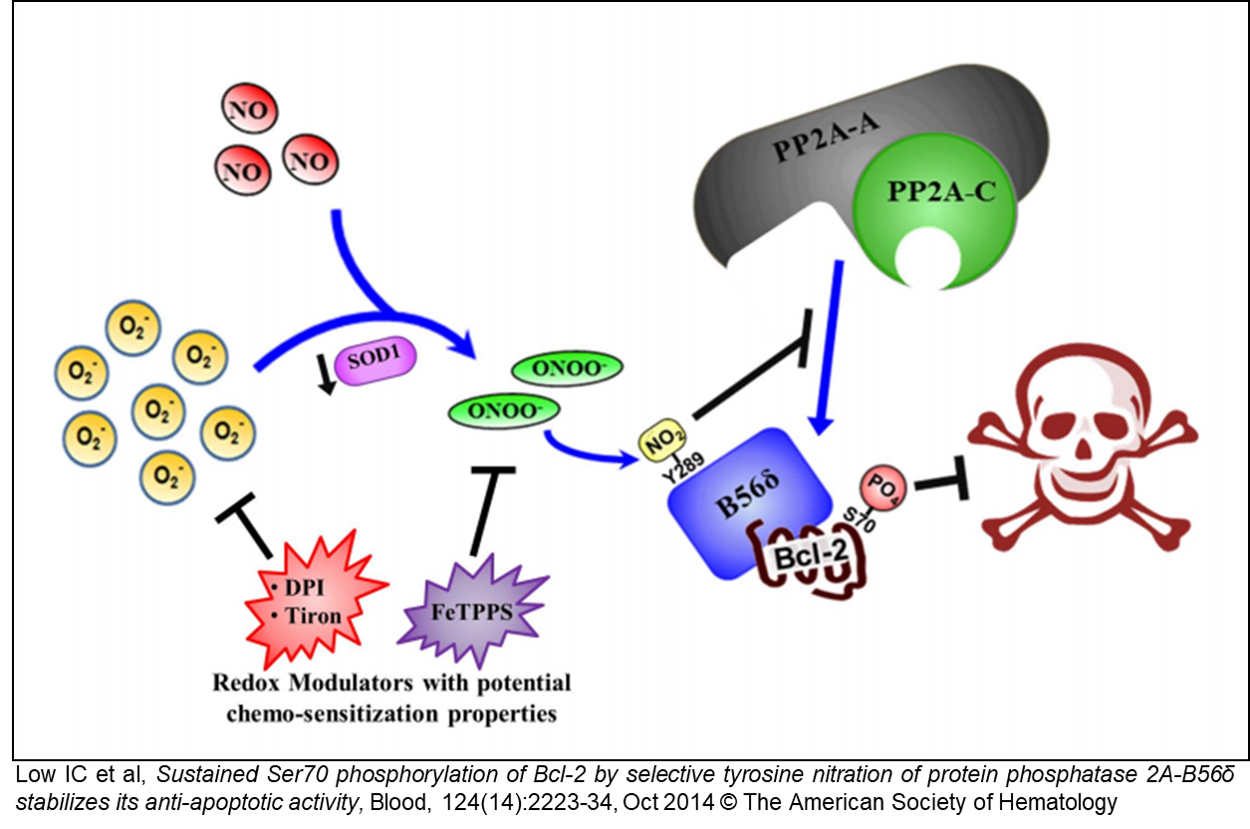
A2. Redox dependent inactivation of tumor suppressor PP2A deregulates Bcl-2-Ser70P
During the course of these studies, we have identified phosphorylation of serine 70 within Bcl-2 as a critical determinant of its redox-dependent pro-survival activity and proceeded to a detailed study of this post-translational regulation of Bcl-2. To that end, we recently identified a novel redox-mediated mechanism of inactivation of the tumor suppressor PP2A. Tyrosine nitration of a specific residue within the B56𝛿 sub-unit was shown to deregulate S70 phosphorylation of Bcl-2, thereby maintaining its phosphorylation and reinforcing its anti-apoptotic activity (Blood, Oct 2014; see also commentary on this work in the same issue). As PP2A is a potent tumor suppressor, and its substrates include tumor suppressors as well as onco-proteins, we are currently investigating the translational relevance of PP2A inactivation, as PP2A regulates a number of important target proteins, including p53, c-Myc and NF-kB.
B: THE DEATH PROMOTING ACTIVITY OF ROS
B1. ROS-mediated sensitization to receptor- and/or mitochondria-mediated apoptosis
While low levels of intracellular ROS promote cell survival, stimuli that induce a robust increase in ROS trigger cell and tissue damage, and death. In this regard, we have demonstrated that small molecule compounds that induce a significant increase in intracellular ROS strongly amplify apoptotic signaling, such as upon ligation of the TRAIL death receptors, via activation of MAPK signaling and/or triggering mitochondrial permeability transition (Cancer Res. 2005; Cell Death Diff. 2007; Cancer Res. 2009; Antiox Redox Sig. 2013; Cell Death Dis. April, June and Oct 2013). During the course of these studies, we have unraveled two interesting observations, (a) identification of a novel protein that is upregulated upon TRAIL-induced apoptosis in a ROS dependent manner, and (b) discovering the activation of surrogate death signaling pathway involving the intermediacy of ROS in a variety of cancer cell types rendered resistant to drug-induced apoptosis. Our current focus and future efforts will be directed at deciphering the molecular mechanism(s) and translational relevance underlying these novel observations.

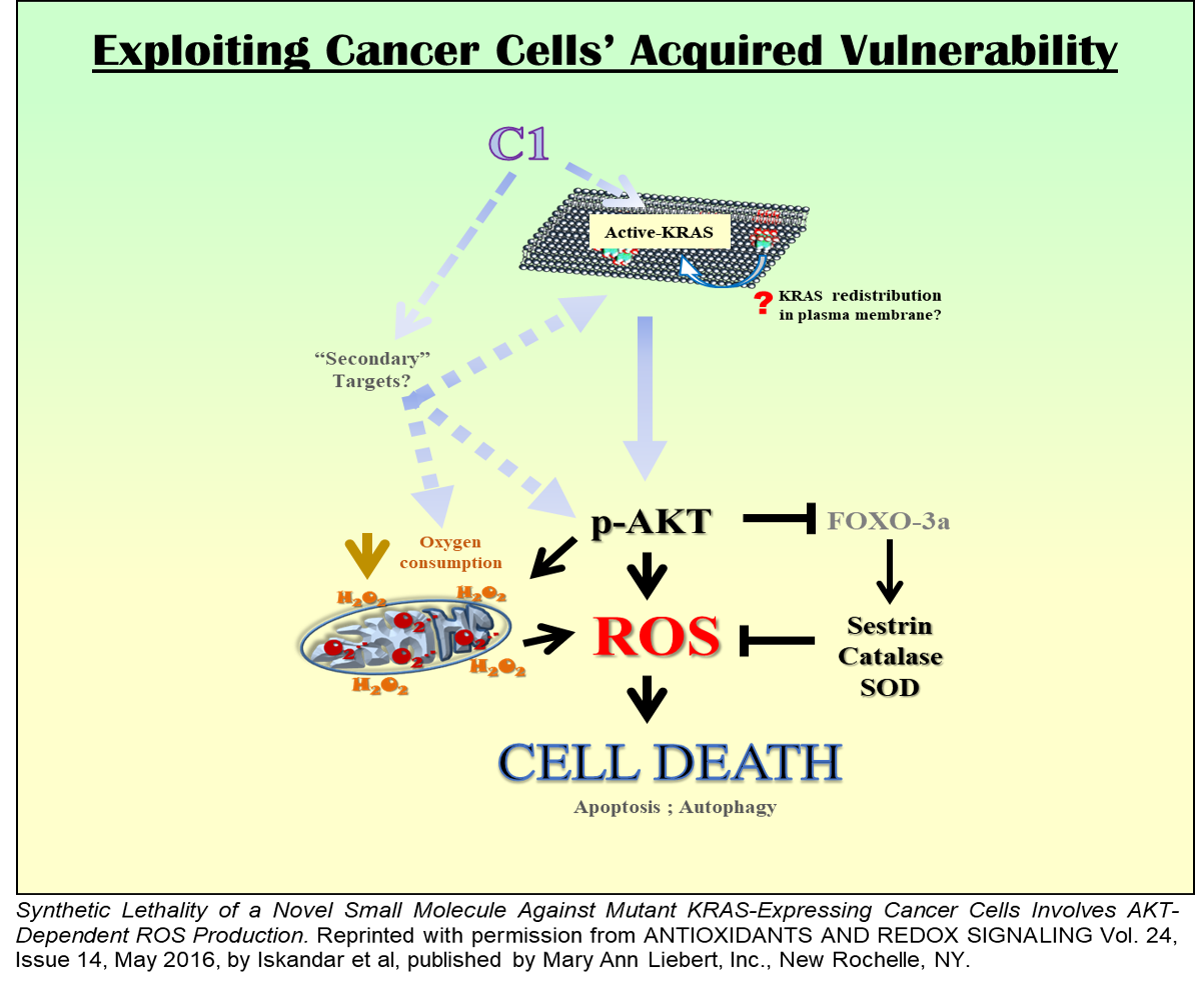
B2. Specific targeting of mutant KRAS driven cancers via ROS-dependent mechanism(s)
Oncogene addiction drives cell survival and proliferation in most cancers. We recently reported the death-inducing activity of a small molecule compound involving ROS-dependent and MAPK-mediated induction of autophagy and apoptosis (PloS One, 2010). By serendipity, investigations into the upstream signaling have revealed a remarkable specificity towards cancer cells expressing mutant KRAS. Intriguingly, the compound activates mutant KRAS, rather than inhibit it, and triggers mutant KRAS driven Akt-mediated mitochondrial redox catastrophe, resulting in selective targeting of these cells (Antiox Redox Sig. 2016). Our ongoing and future studies will be focused on identifying the mechanism of specific activation of mutant KRAS and the involvement of the PI3K signaling axis in the phenotype of cell death triggered by the small molecule compound. Considering the relative dearth of mutant KRAS targeting agents, these findings could have tremendous therapeutic potential for developing redox-based strategies to render cancer cells susceptible to their addiction.