
Research Interests
We employ a multidimensional tool set to investigate mechanisms governing formation and dysfunction of synapses and neuronal circuits. Currently, our efforts focus on the main areas: listed below.
The role of FEZ1 in the establishment and maintenance of neuronal networks
The establishment of synapses in developing neurons is a major logistical undertaking involving the systematic transport of thousands of synaptic proteins by motor proteins along microtubule-based cytoskeletal highways (Yagensky et al., 2016). These proteins must be carefully sorted and routed to their specific destinations to support the rapid growth and expansion of nascent synaptic sites. Motor-specific adapters play crucial roles in this process by conferring motors not only with the means to selectively recognize and bind cargoes, but also as molecular scaffolds to recruit various signalling pathways that modulate the activity of motor complexes.
We uncovered a major pathway for synaptic protein transport mediated by the Kinesin-1 adapter fasciculation and elongation protein zeta-1, FEZ1, which is selectively expressed in neurons (Figure 1). We showed that many key active zone proteins as well as synaptic vesicle proteins are present on FEZ1 transport vesicles (Chua et al., 2012; Butkevich et al., 2016). Significantly, disruption of FEZ1 function in neurons causes a dramatic reduction of synapses and is accompanied by a massive accumulation of synaptic cargoes in axons. Moreover, FEZ1-null neurons show significantly impaired axo-dendritic development and were no longer unresponsive to stimulation by Netrin-1 and Sema3A guidance cues (Chua et al., 2021).
Remarkably, loss of FEZ1 also affected the development of human motor neurons (Gunaseelan and Wang et al., 2021). Importantly, knockdown of FEZ1 in Drosophila causes locomotion defects that recapitulate motor impairments detected in Jacobsen syndrome patients, where the FEZ1 gene is frequently deleted. Collectively, these findings demonstrate the critical role of FEZ1 in establishing neuronal networks in the central as well as peripheral nervous systems.
Current efforts focus on elucidating how FEZ1 interfaces with various intracellular signalling pathways to influence the neuronal network function.
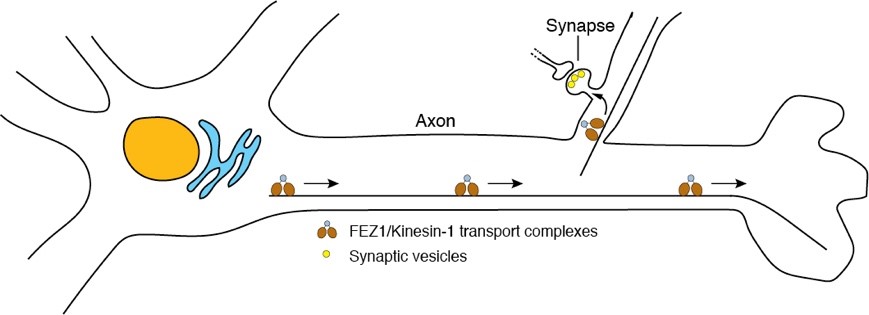
Figure 1. Synaptic protein transport by FEZ1 plays critical roles in synapse formation and function.
Human brain organoids and human stem cell-derived neurons as models to study role human brain development and neurodevelopmental disorders
A critical step in the development of the human brain is the systematic organisation and wiring of the estimated 100 billion neurons into functional neuronal networks essential for its optimal function. In humans, establishment of these networks occur progressively over the first two decades of life. Neuronal synapses critically control the flow of information across neuronal networks. Synaptic failure leading to the alteration or even the destruction of neuronal networks is a critical event underlying neurodevelopmental and neurodegenerative disorders. Yet, despite the explosion of knowledge regarding key proteins and processes governing synaptic function in the preceding decades, much remains to be understood about the organizing principles of synapses and the rules governing the establishment and maintenance of synaptic networks during human brain development. Importantly, a growing number of synaptic proteins have been identified as risk factors in major neuropsychiatric and neurological disorders.
To elucidate how neuropsychiatric risk genes contribute to human brain development and neurodevelopmental disorders, we have successfully established human cerebral brain organoids (CBO) and human monolayer neurons using human embryonic stem cells (hESCs) or induced pluripotent stem cells (iPSCs) (Figure 2). These platforms serve not only as bridges to test and validate data generated from model organisms to humans but will also allow us to directly examine the effects of these perturbations in a system closely recapitulating that of human brain development in vivo.
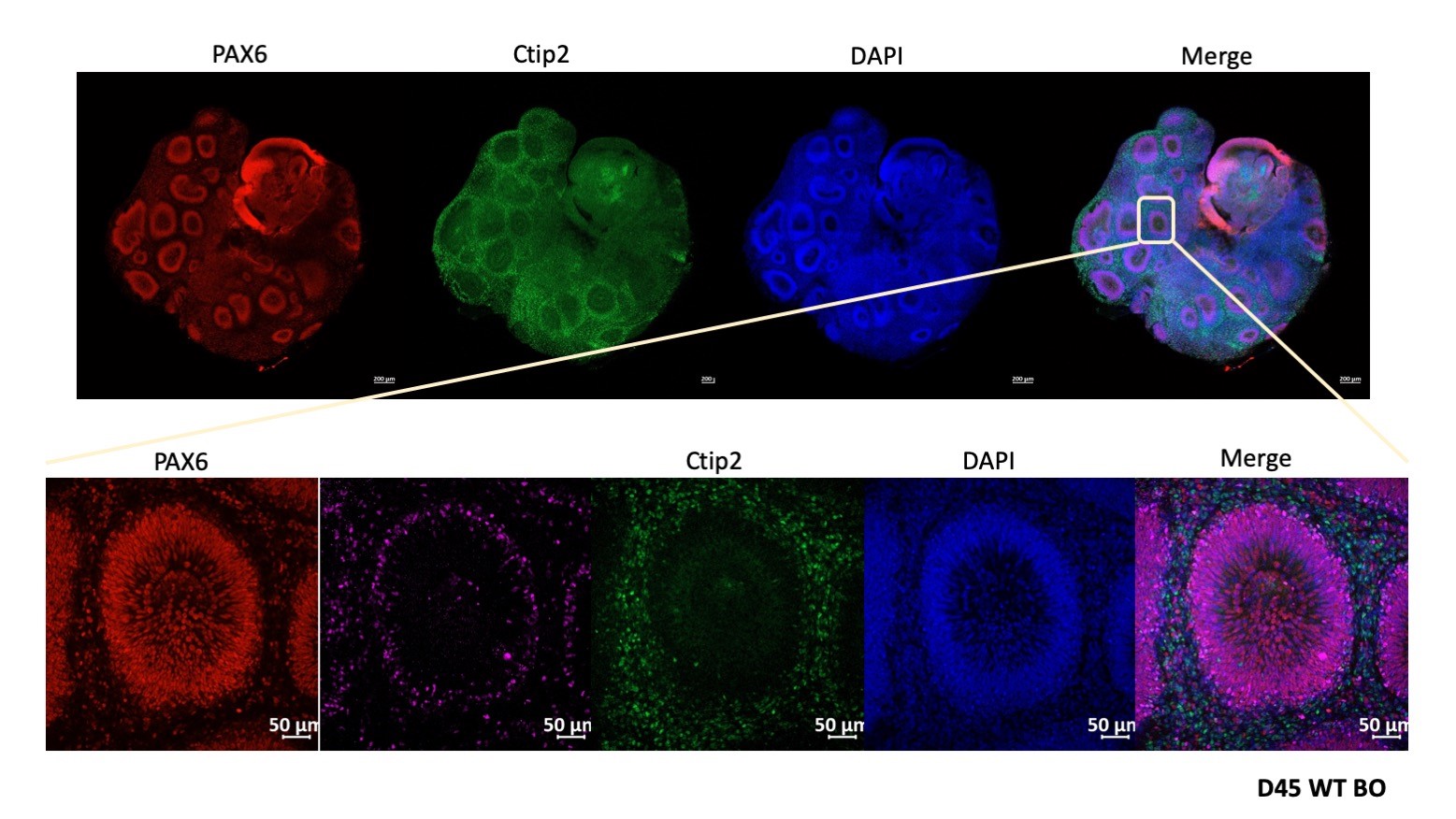
Figure 2. Human cerebral brain organoids to model early human brain development.
We employ CRISPR-Cas9 gene-editing to introduced mutations in stem cell lines to manipulate the expression of selected risk genes and derive brain organoids and monolayer neurons from these cells. These organoids are characterized using a battery of assays including tissue clearing, immunohistochemistry, RNAseq and immunoblotting to profile morphological and gene expression changes. Collectively, the efforts aim towards identifying the molecular mechanisms involved in early brain development and neurodevelopmental disorders.
Molecular mechanisms of neuronal network degeneration
Disruption of neural circuits either via synapse loss from individual neurons or of entire neurons themselves form the basis of major neurological disorders, including Alzheimer’s disease (AD). Our work has highlighted that FEZ1-mediated synaptic transport is disrupted in an AD transgenic mouse model, presumably via dysregulated signalling through microtubule-affinity regulating kinases (MARKs, a group of tau kinases) that phosphorylate and activate FEZ1 (Butkevich et al., 2016). Aggregates of FEZ1 and its synaptic cargoes progressively accumulate during disease progression in the brains of these animals. These findings shed further light on how disrupted axonal trafficking, already implicated in AD and other major neurodegenerative disorders, contributes to neuropathology in these disorders. Ongoing efforts aim to understand the relationship between how pathogenic Aβ species, acting on signaling mechanisms regulating FEZ1 function, disrupt axonal transport to cause synapse loss and neuronal death.
In parallel, by temporal profiling of proteome changes during the course of disease progression in 3Tg-AD mice, we identified a number of stage-specific AD markers (Yagensky et al., 2019). In particular, we identify Hebp1 as a marker that is elevated at the asymptomatic phase in transgenic mice that is also highly elevated in the brains of patients diagnosed with rapidly progressing forms of AD. We show that the protein, which binds heme, causes mitochondrial-mediated apoptotic neuronal cell death upon exposure of neurons to elevated levels of heme and/or A42. This pathway contributes to the progressive loss of neurons and disrupted neural circuitry which constitutes the major neuropathological process in AD (Figure 3).
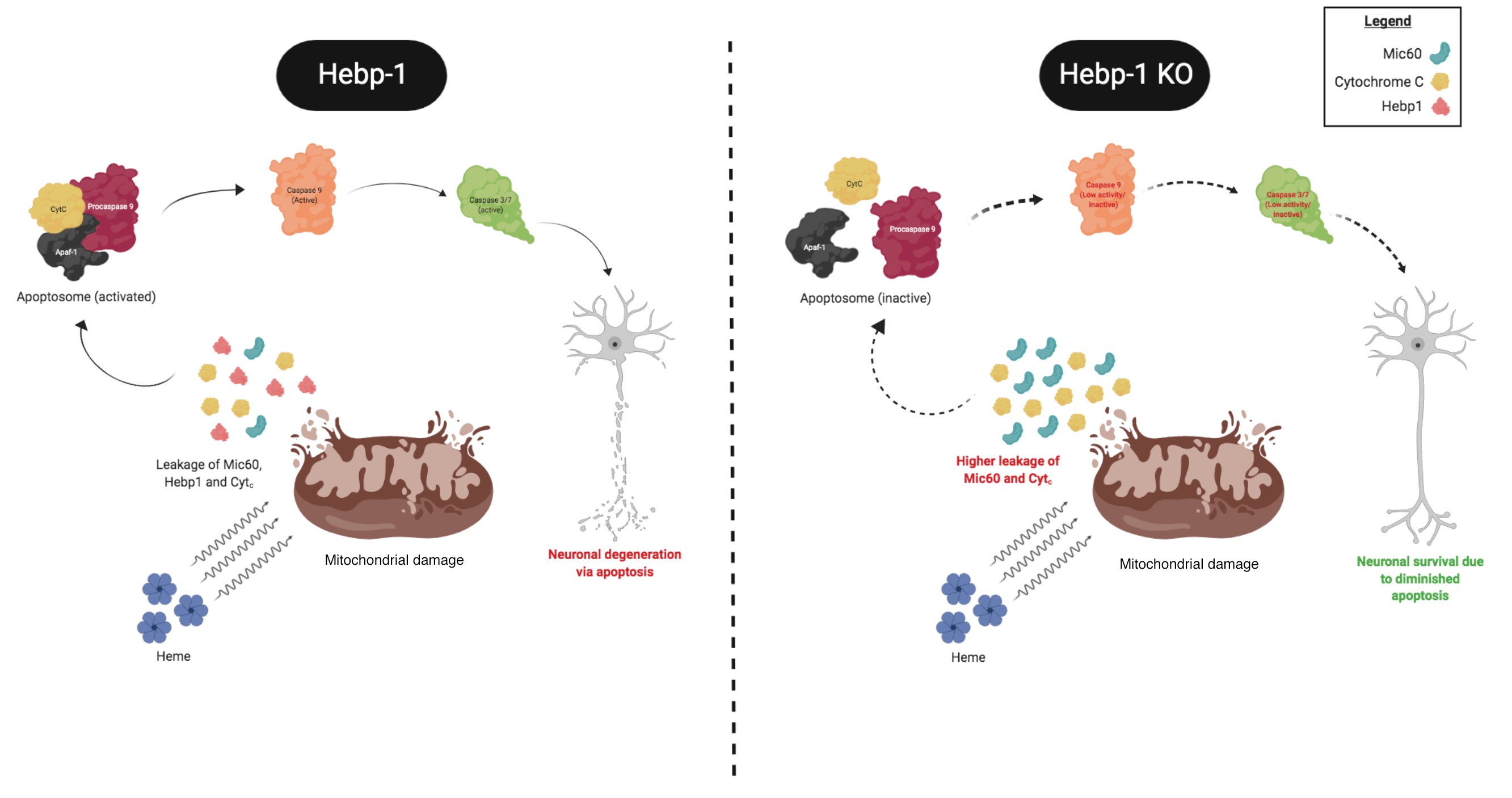
Figure 3. The involvement of Hebp1 in early AD neuropathogenesis. (Artwork by Saravanan)
These findings are significant given that accumulation of Aβ within brain vasculature results in Cerebral Amyloid Angiopathy, wherein microvessel damage results in leakage of free heme into brain tissue. Thus, Hebp1-mediated neuronal cell death would also be of relevance to other neurological disorders associated with vascular damage (e.g. vascular dementia and stroke). We are currently investigating how Hebp1 triggers activation of the apoptotic cascade in neurons and how, as a mitochondria-associate protein, it contributes to mitochondrial function in this capacity. Concurrently, we are evaluating the efficacy of Hebp1 as an asymptomatic AD biomarker to identify AD patients even before they exhibit signs of mild cognitive impairment. Also, we are examining if Hebp1 could contribute to the neuropathogenesis of stroke and vascular dementia. We are also planning to validate the other markers identified in this study.