

Issue 39 / August 2021
Science of Life
Found – A Key Protein Essential for Building the Brain’s Neuronal Network
hose of you who might have been mischievous enough at some juncture to tinker around electrical wiring will not forget the jolt of electricity coursing through your finger as a result of accidental miswiring. Almost always, the sensation immediately triggers lively hopping around but the effect is fortunately transient and leaves little to no lasting repercussion.
Now, can you imagine what could happen if neuronal networks in our brain get miswired?
To understand this, one should first appreciate that neurons, the functional workhorses of our brain, are organised into functional cellular networks—but neurons do not begin life as part of a network. Analogous to how a circuit board is created, immature neurons derived from neuronal precursors must send out cellular projections to make connections (synapses) with their designated target neurons to form the various neuronal networks that endow us with the means to sense, respond and permanently remember, for instance, that electric jolt.
Growing neuronal networks
For the sake of this article, and at the expense of some oversimplification, I highlight two requisite processes for neuronal networks to form. Firstly, growing neuronal projections must know where to go. Secondly, growth of the projections must be supported by a continual supply of biological raw materials.
The first process is accomplished via signals comprising chemical gradients secreted by target cells, known as guidance cues, that are picked up by receptors located at the tips of the growing projections (growth cones) (Figure 1A). During brain development, different neurons project to different areas of the brain. Complex arrays of guidance cue gradients generated across different brain axes ensure that billions of neurons are correctly wired up with each other to form both short- and long-range networks. Major guidance pathways involved in this process include guidance cues such as Netrins and Semaphorins and their associated receptors. Thus, migrating neuronal projections utilise guidance cue receptors located at their growth cones to sense Netrin and Semaphorin gradients originating from the target cell regions (Figure 1A). Depending on which guidance cues are detected and the cell types they bind to, neuronal projections are then either steered (attracted) toward their targets or repelled from incorrect ones.
FEZ 1: The protein propelling healthy neuron growth
Now that they know where to go, these projections need to grow towards their targets to form connections (synapses). As mentioned earlier, growing projections require a constant delivery of new biological materials (also known as cargoes) to support their elongation (Figure 1A, inset). Enter the Kinesins, the biological motors responsible for transporting cargoes within cells. Although motors supply the driving force to move cargoes around, they also require additional proteins that enable them to recognise and carry their respective cargoes and to regulate transport. One such protein that is essential for both functions is fasciculation and elongation protein zeta 1 (FEZ1). Binding of FEZ1 to Kinesin regulates its activities and allows the motor to bind and transport their cargoes. We and others have shown that FEZ1 mutations severely disrupt intracellular transport and cause massive traffic jams in neuronal projections. Incidentally, these abnormalities are associated with neurodegenerative disorders, such as Alzheimer’s disease.
As with any viable business model, a good intermediary is required to ensure that supply meets demand. Thus, while it would seem commonsensical that guidance cues should liaise with the intracellular machinery to direct cargoes solely to regions of growth as neuronal networks form, how this is achieved and the identity of the intermediary connecting the two processes has hitherto been little known.
Our recent work1 provides an important illumination regarding this knowledge gap by identifying FEZ1 as the intermediary in question. As mentioned earlier, attractive guidance cues such as Netrin-1 and Semaphorin 3a steer growing projections towards their neuronal targets. Remarkably, we observe that deleting FEZ1 expression in neurons not only blocked the ability of guidance cues to stimulate the growth of neuronal projections, it also severely stunted their growth (Figure 2)! These observations uncover miswiring events that occur in the absence of FEZ1, which is expected to disrupt the resultant neuronal network formed (Figure 1B).
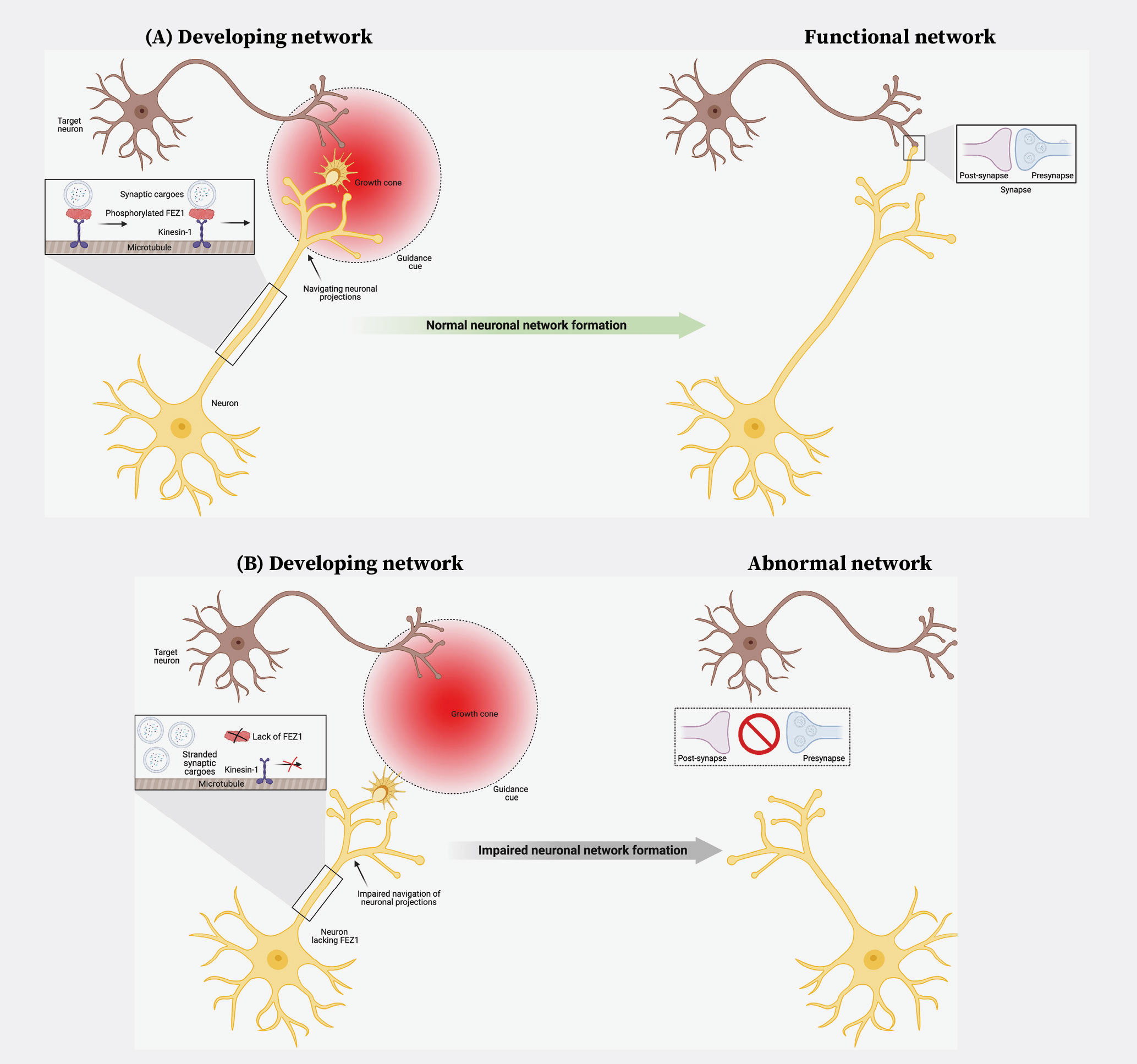
Figure 1. (A) Formation of neuronal networks during brain development requires guidance cues to help developing neuronal projections migrate to their neuronal targets. Upon contact, the projections form synapses to allow communication between the neurons. (B) Loss of FEZ1 disrupts communication between guidance cue signalling and the intracellular transport pathway. This, in turn, affects neuronal network formation during brain development.
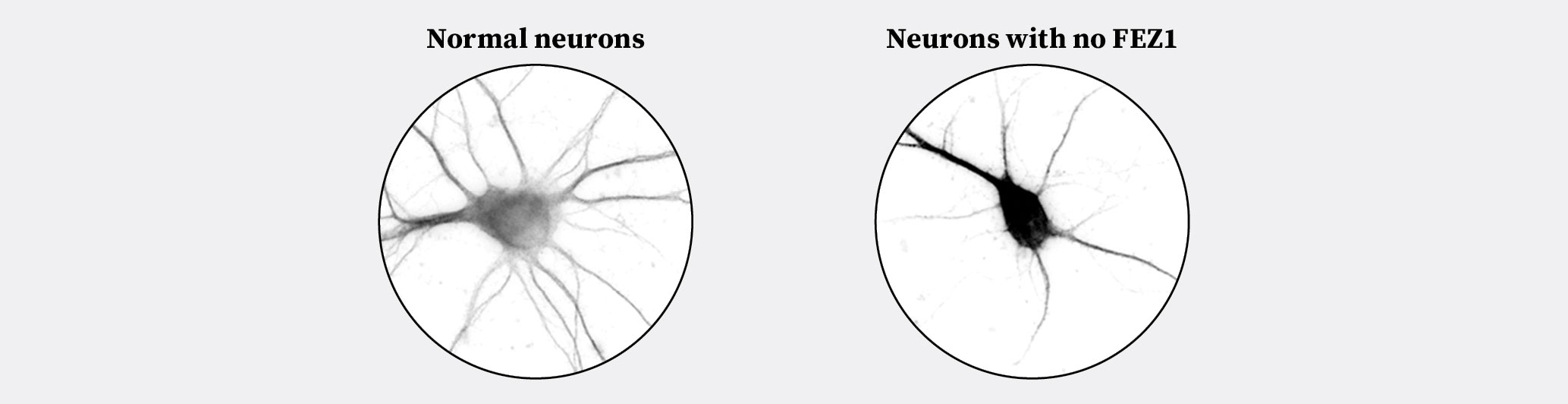
Figure 2. Left panel: normal neurons show proper development of neuronal projections. Right panel: neurons without FEZ1 show noticeably fewer and shorter projections reflecting abnormal development. Images modified from Chua et al., 2021.
Missing FEZ1 linked to mental health disorders
Significantly, neuronal network disruptions are thought to cause major mental health disorders (Figure 3). FEZ1 itself has been implicated in at least schizophrenia and attention deficit hyperactivity disorder (ADHD). The gene encoding FEZ1 is also frequently deleted in patients affected by Jacobsen Syndrome (JS), a rare chromosomal disorder where the terminal region of Chromosome 11q is deleted. Remarkably, behavioural abnormalities (related to ADHD, autism spectrum disorder (ASD) and, less frequently, schizophrenia) and cognitive deficits have been documented in JS patients, adding support to the notion that abnormalities in FEZ1 contribute to such disorders. These patients also exhibit psychomotor impairments, including gross and fine motor delays. Of note, we recently demonstrated that FEZ1 deletion also causes delays in human motor neuron development2. This indicates that its involvement in neuronal network formation extends beyond the central nervous system to participate in the formation of motor circuits.
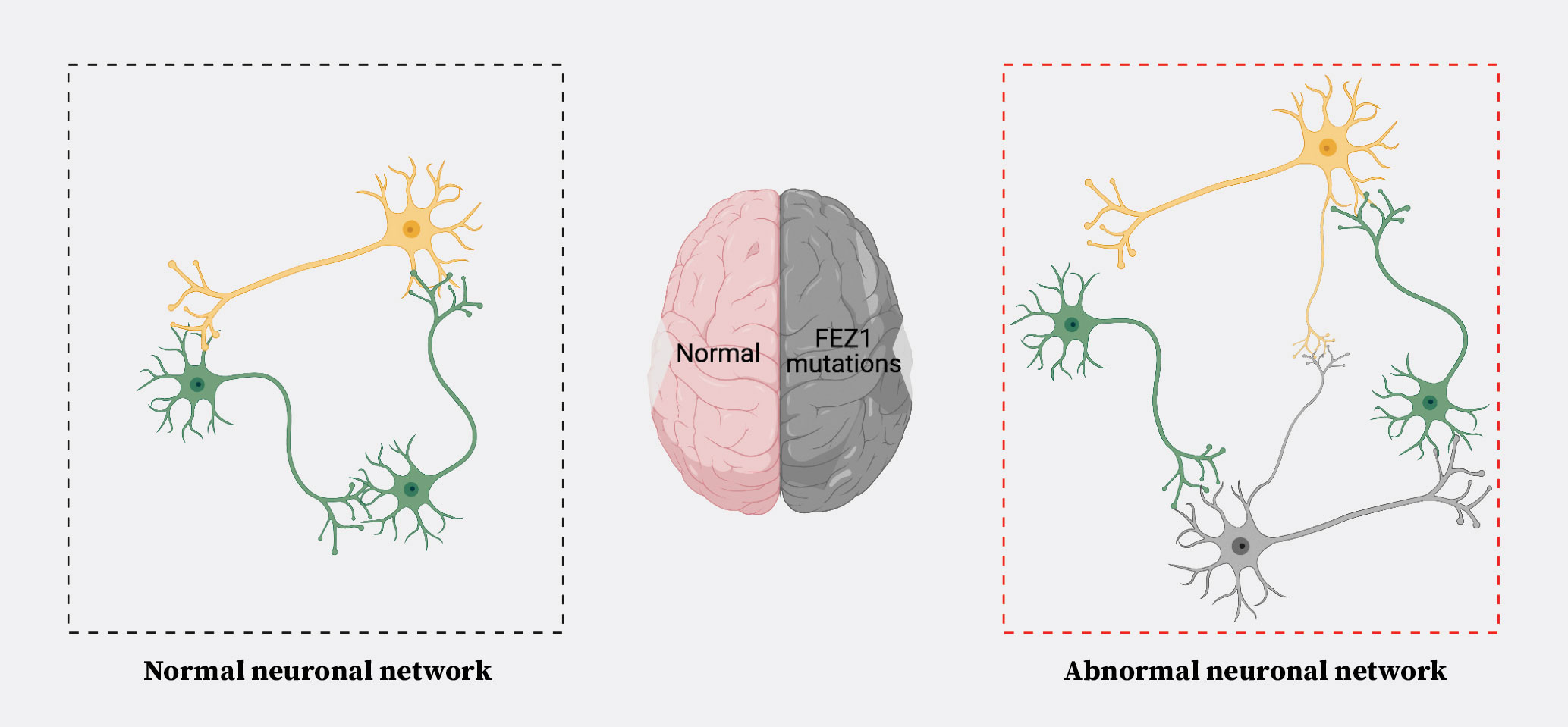
Figure 3. Alternation of neuronal networks as an importance cause of mental health disorders.
Moreover, FEZ1 is involved in another important step of neuronal network formation–the formation of synapses (Figure 1A). We previously reported that loss of FEZ1 in neurons dramatically reduced the number of synapses (i.e., the number of connections formed between neurons). Furthermore, FEZ1-deficient flies show abnormalities in their neuromuscular junctions (synapses formed between motor neurons and muscles) that correlated with movement disorders observed in these flies. These findings identify another layer of alteration to neuronal networks when FEZ1 function is compromised or lost.
Collectively, our findings highlight FEZ1 as a critical player in the formation of central as well as peripheral neuronal networks. It does so by acting as a convergence point to integrate key pathways involved in neuronal development. Elucidating FEZ1’s functions uncovers but the tip of the iceberg in our efforts to define the complex and polygenic causes of mental health disorders. Building upon these efforts, we will progressively unravel and demystify the layers of complexity surrounding the origins of these disorders that will allow eventual development of targeted strategies to treat affected individuals.
-
Jie Yin Chua, Shi Jun Ng, Oleksandr Yagensky, Erich E. Wanker, John Jia En Chua, FEZ1 Forms Complexes with CRMP1 and DCC to Regulate Axon and Dendrite Development, eNeuro 26 March 2021, 8 (2) ENEURO.0193-20.2021; DOI: 10.1523/ENEURO.0193-20.2021.
-
Saravanan Gunaseelan, Ziyin Wang, Venetia Kok Jing Tong, Sylvester Wong Shu Ming, Rafhanah Banu Bte Abdul Razar, Sumitra Srimasorn, Wei-Yi Ong, Kah-Leong Lim, John Jia En Chua, Loss of FEZ1, a gene deleted in Jacobsen syndrome, causes locomotion defects and early mortality by impairing motor neuron development, Human Molecular Genetics, Volume 30, Issue 1, 1 January 2021, Pages 5–20, https://doi.org/10.1093/hmg/ddaa281.
Illustrations by Saravanan Gunaseelan, Research Fellow, Department of Physiology, NUS Medicine.


