

Issue 47
Aug 2023
A WORLD IN A GRAIN OF SAND
by Associate Professor Vincent Chow, Department of Microbiology and Immunology, NUS, Professor T Narasaraju, Adichunchanagiri, Institute of Medical Sciences, India
Due to its direct contact with the external environment and continuous exposure to various microbial pathogens, the respiratory system constitutes a primary target for viral, bacterial, and other infections. Respiratory infections account for about one-third of deaths arising from infectious diseases worldwide, and pneumonia is the second principal cause of mortality in Singapore1. The recent COVID-19 pandemic highlighted the devastating impact of respiratory infections on global human health and economic activity. Other respiratory viruses such as influenza continue to pose significant pandemic threat. Furthermore, certain primary viral infections such as influenza often predispose to secondary bacterial or other infections in the lungs which can potentially aggravate the disease outcome.
Respiratory pathogens can obviously directly damage lung tissues by their sheer replication and virulence factors, which can be managed by antimicrobial therapy. However, exaggerated inflammation or immunopathology may also inflict “collateral damage” to host tissues.
Neutrophils: First responders of lung innate immune response
The lung innate immune response is unique and well-organised as it is constantly exposed to innumerable microbial pathogens. The early host innate immune regulation is mediated by cells (including neutrophils, macrophages, natural killer cells, dendritic cells) which act as first lines of defense in eliminating microbes via recognition of pathogen-associated molecular patterns (PAMPs) present in microbes. The innate immune cells express surface molecules called pathogen recognition receptors (PRRs) that specifically recognise PAMPs.
The influx of neutrophils is recognised as a hallmark of early host defense—they are the most abundant leukocytes in the circulation, and act as rapid responders to infection or injury. Neutrophils are short-lived and differentiated innate immune cells that exert phagocytic activity to clear microbes or infected cells. Neutrophils contain cytoplasmic granules that produce reactive oxygen radicals and secrete various toxic enzymes such as neutrophil elastase (NE), myeloperoxidase (MPO), and matrix metalloproteinases (MMPs), which aid in microbial killing. Once released from the bone marrow, neutrophils mature and circulate for a few days as active phagocytic cells. In response to myriad external stimuli such as infection, neutrophils undergo degranulation to release toxic granule enzymes such as NE, MPO and MMPs2. Besides their phagocytic function, neutrophils also undergo a specific cell death mechanism known as NETosis. During NETosis, neutrophils and their nuclei rupture and release their chromatin fibers outside the cell to form neutrophil extracellular traps or NETs3. The chromatin strands in the NETs contain DNA, histone proteins, and toxic granule proteins. Although neutrophils and NETs contribute to host immunity, excessive neutrophil infiltration and persistent release of NETs may aggravate host tissue damage, leading to organ failure and even death in several disease conditions including pneumonia.
Friends, foes or “frenemies”?
Neutrophil recruitment and recognition of bacterial pathogens are key elements of host defense against bacterial infection. Recognition of bacterial pathogens is mediated by PRRs that bind to bacterial PAMPs to enhance phagocytosis and induce release of cytokines and chemokines (such as IL-8, MIP-2, IL-6, IL-1β), which in turn trigger inflammatory cellular influx at the site of infection. The PRR-PAMP ligation together with serum complement enhance bacteria-ingested phagosome formation. The phagosome-mediated bactericidal effects involve free-radical-generating enzymes (including MPO, superoxide dismutase, NADPH oxidase) which generate reactive oxygen intermediates (such as superoxide, hydrogen peroxide, hypochloric acid) that kill bacteria. Neutrophils also kill bacteria by an oxygen-independent mechanism via release of toxic microbicidal agents (such as elastase, proteinase-3, cathepsins, defensins, lysozyme) that kill ingested bacteria2.
The release of NETs during bacterial infection was initially believed to trap and kill bacteria in the extracellular environment3. However, subsequent evidence revealed that the released NETs can trap bacteria to exert bacteriostatic, but not bactericidal effects. NETs are capable of trapping certain bacteria without killing them. Certain bacteria have evolved strategies to inhibit NETs via synthesis of microbial DNase enzymes that can digest NETs. NETs released during bacterial infection cause collateral host tissue damage by exacerbating inflammatory responses. Studies from our laboratory demonstrated that NETs released in response to influenza infection do not confer killing effect against bacterial pathogens. We found that secondary lung infection with Streptococcus pneumoniae (the pneumococcus) following primary influenza incites widespread NETs to inflict extensive tissue destruction and death, thus indicating the detrimental role of NETs in exacerbating inflammation4,5.
Potential pathologic “foes”: Excessive neutrophils and NETs in viral pneumonia
Evidence is accumulating to implicate the deleterious effects of excessive neutrophils and NETs in mediating acute lung injury caused by viral pathogens. Our laboratory provided early evidence that excessive pulmonary sequestration of neutrophils contributes to alveolitis and can exacerbate pulmonary pathology in severe influenza pneumonia6. We observed that NETs released in influenza-infected lungs were entangled with alveolar epithelial cells as well as endothelial cells in the pulmonary vasculature, thus disrupting the alveolar-capillary barrier in mice that succumbed to fatal influenza pneumonia. Enhanced NETs induction correlated with severe hypoxemia, thus supporting the phenomenon of aggravated lung tissue damage by NETs. Among various toxic proteins elaborated by NETs, histone proteins exert highly cytotoxic and coagulative effects. Histones released into the extracellular space bind to platelets and instigate platelet aggregation; and purified histones can induce microvascular thrombosis in mice. Indeed, increased histone levels are detected in nasal wash samples of influenza-infected patients—thus, elevated histone levels (e.g. citrullinated histone H3) may serve as a clinical marker of pulmonary injury7. Increasing evidence suggests that accumulated NETs disperse toxic nuclear histone proteins as well as granule proteins (such as MPO, NE, MMPs) which can disrupt the alveolar epithelial-capillary barrier, degrade extracellular matrix proteins, and trigger severe vasculitis in influenza pneumonia (see Figure).
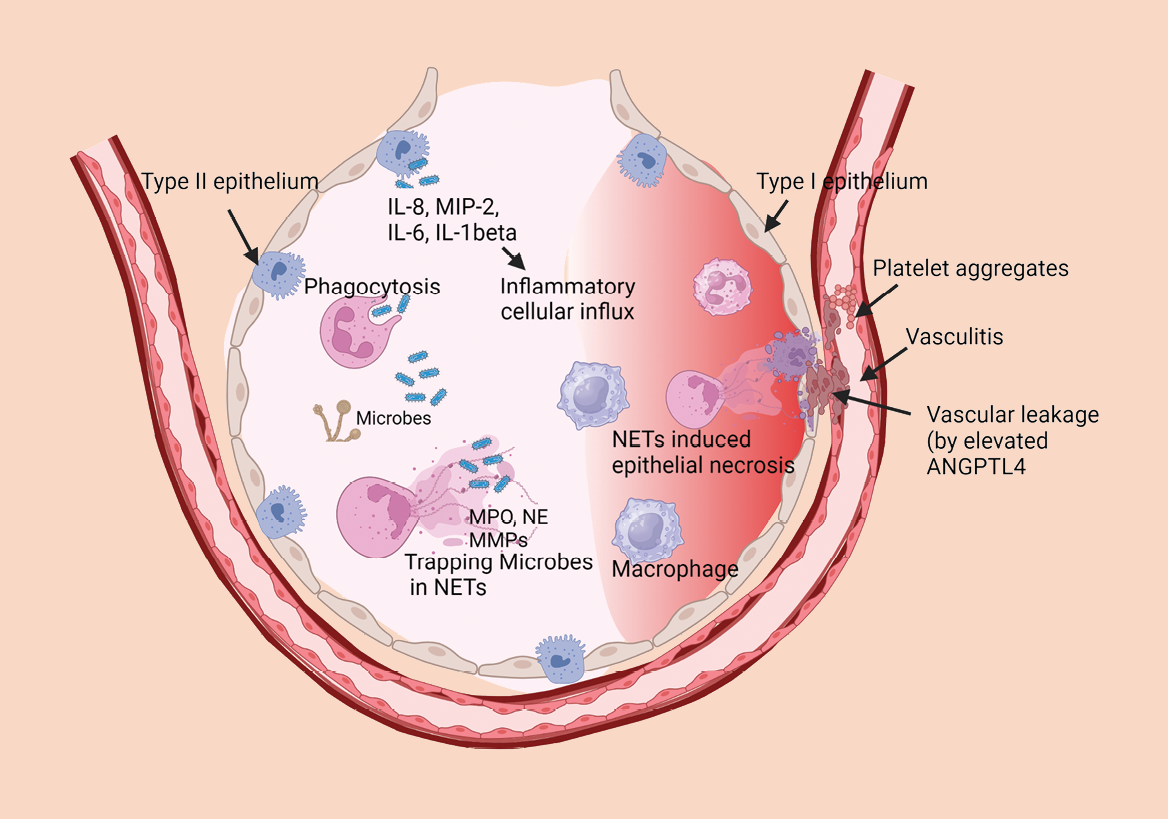
Neutrophils and neutrophil extracellular traps (NETs) in pneumonia: Too much of a good thing?
More recently, the role of neutrophils and NETs in SARS-CoV-2 infection attracted considerable attention8, given that COVID-19 patients exhibited a progressive rise in neutrophilia in non-survivors compared with survivors with mild to moderate disease. Heightened NETs levels were observed in severely ill COVID-19 patients, alluding that NETs could serve as a potential clinical marker of tissue injury in COVID-19. Lung autopsies revealed severe pulmonary sequestration of neutrophils and extensive release of NETs in the lower respiratory tract in fatal COVID-199. Thrombotic lesions were commonly reported in critically ill COVID-19 patients associated with significant mortality, especially those with complications of coronary failure, stroke, deep vein thrombosis, and pulmonary embolism. In support of this, lung autopsies of COVID-19 patients displayed immune-related thrombosis with widespread occlusion of small blood vessels by aggregated NETs. Interestingly, neutrophil-platelet aggregates (NPAs) were also augmented in patients with severe COVID-1910. We have previously documented that NPAs induce thrombosis and NETosis in fatal influenza pneumonia in mice11. The enhanced accumulation of NETs also inflicts direct endothelial necrosis, thus leading to vascular injury and leakage. Several studies also reported diffuse alveolar damage, abnormal lung tissue remodeling, fibrosis, dysregulated neutrophil activity, and extensive accumulation of NETs that correlate with COVID-19 severity12.
Another important pathogenic mechanism of pneumonia is mediated by the angiopoietin-like 4 or ANGPTL4 protein, elevated levels of which can culminate in increased vascular permeability and extravasation of fluid into lung tissues which may ultimately lead to potentially life-threatening acute respiratory distress syndrome13,14,15.
Conclusions and potential applications
The discovery of NETosis has opened new avenues in better understanding the pathophysiology of respiratory infectious diseases, especially pneumonia. Although comprising a critical part of innate immune defense, NETs may potentially inflict harmful “bystander” effects on the host. Pre-clinical studies and clinical trials have been initiated to target NETosis in acute respiratory infections such as COVID-19 and influenza. For example, drugs such as DNase-I that can digest NETs are being investigated. However, it is unclear how the NETs will be removed from the infection site, as they are highly fragile and can diffuse into the deeper lungs as well as systemically. The real challenge is that many components of NETs (such as histone proteins, NE, MPO, MMPs) can induce significant inflammatory responses even after they are separated from NETs. Hence, drugs that target the upstream signaling processes for formation of NETs or components of NETs could be more feasible to prevent or ameliorate NETs-mediated tissue injury in respiratory infections. Examples include deploying potent anti-inflammatory corticosteroids such as dexamethasone16; targeting the neutrophil CXC chemokine receptor 2 (CXCR2) to suppress neutrophilic inflammation17; or targeting platelet activation by using clopidogrel to mitigate formation of NPAs11.
Singapore Medical Journal 2018; 59:190–198.
Nature Reviews Immunology 2013; 13:159–175.
Science 2004; 303:1532–1535.
Frontiers in Immunology 2013; 4:56.
Oncotarget 2016; 7:19327–19340.
American Journal of Pathology 2011; 179:199–210.
American Journal of Pathology 2018; 188:135–148.
Frontiers in Pharmacology 2020; 11:870.
Journal of Infectious Diseases 2021; 223:1512–1521.
Thrombosis & Haemostasis 2020; 120:1733–1735.
American Journal of Respiratory Cell & Molecular Biology 2019; 61:689–701.
Respiratory Research 2021; 22:32.
Cell Reports 2015; 10:654–663.
mBio 2019; 10:e02469-18.
Critical Care Explorations 2023; 5:e0827.
Frontiers in Immunology 2017; 8:60.
American Journal of Pathology 2021; 191:669–685.
More from this issue

OFFICE FOR STUDENTS
Community Service Programme Gives Students Practical Learning Opportunities
SCIENCE OF LIFE
Novel Cancer Therapy Prolongs Lives of Terminally Ill Dogs
