
Dr Chen Zhixiong with Ms Choo Zhang’e, first author of the paper
Story by Dr Khor Ing Wei and Cher Boon Meng
Neuroblastoma is a rare but aggressive cancer that affects immature nerve cells in the sympathetic nervous system in infants and young children. The system comprises nerve cells and fibres that control body functions such as heart rate and digestion.
Currently, neuroblastoma is usually treated with cytotoxic agents that kill the cancer cells but are not personalised for the specific tumours in an individual patient. To help address this, the roles of several genetic changes in neuroblastoma tumours are being deciphered. Researchers had previously found that deleting one of these genes, KIF1B, results in an especially aggressive type of cancer with a very poor prognosis.
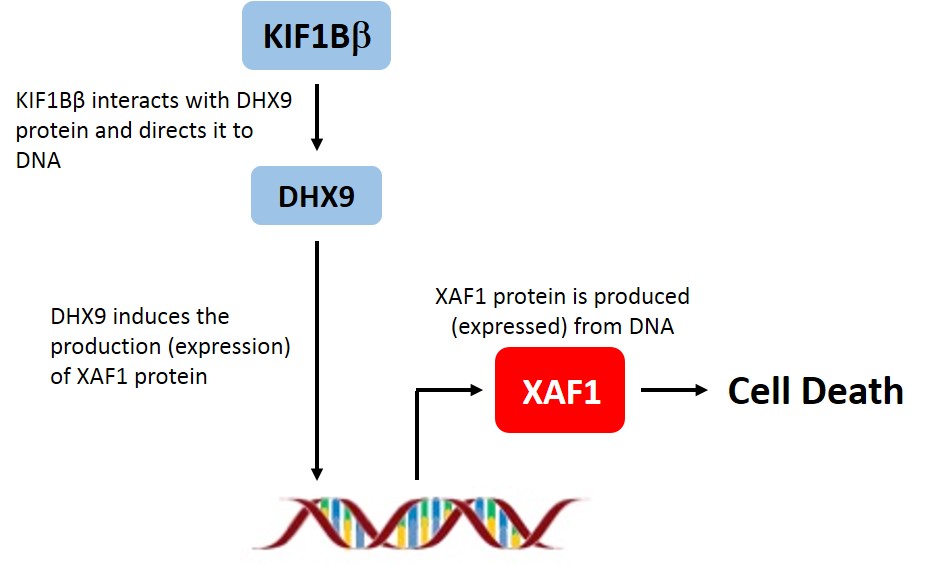
Intrigued, an international team of researchers set out to understand what happened when KIF1B is deleted, or absent. Dr Chen Zhixiong of the Department of Physiology at NUS Medicine, Dr Kenneth Chang, Dr Amos Loh and Dr Soh Shui Yen from the KK Women’s and Children’s Hospital, along with researchers at the Ludwig Institute for Cancer Research in Switzerland and the Moffitt Cancer Center in the U.S. examined neuroblastoma cell lines, animal models, and tumours from neuroblastoma patients and found that a form of KIF1B known as KIF1Bβ was responsible for the expression of another protein called XAF1.
When the researchers suppressed the expression of XAF1 in neuroblastoma cells, more tumour cells survived. Similarly, when XAF1 expression was suppressed in animal models, tumour growth increased dramatically over that found in control mice. Doing the reverse and increasing the amount of XAF1 caused the tumours to shrink. Going a step further, the research team showed that the absence of XAF1 was responsible for the aggressive tumour growth in neuroblastoma patients with the KIF1B gene deletion. This discovery explained why the tumours in these patients grew so uncontrollably.
After they had uncovered the tumour suppressive effect of XAF1 in cells and mouse models, the team moved to determine whether the same effect would be seen in human tumours. As they expected, the level of XAF1 corresponded with the 5-year survival rate for patients. Patients with tumours which had higher levels of XAF1 had a higher survival rate (87%) than those with tumours which had low or no XAF1 (70%).
Equally striking, almost all of the survivors who showed evidence of disease had low or no XAF1, suggesting that XAF1 may also correspond to the quality of survival. Their findings were published in Oncotarget in April this year.
The work elegantly demonstrates that XAF1 is a tumour suppressor involved in neuroblastoma. Besides holding promise as a prognostic marker for neuroblastoma, XAF1 could also be a target for new treatments to be tested in pre-clinical models that are currently being developed in conjunction with the KK Women’s and Children’s Hospital. Treatments that restore XAF1 expression or mimic its function could be effective for this rare but devastating childhood cancer.
Reference:
Choo Z, Koh RY, Wallis K, Koh TJ, Kuick CH, Sobrado V, et al. XAF1 promotes neuroblastoma tumour suppression and is required for KIF1Bβ-mediated apoptosis. Oncotarget. 2016 Apr 15. Epub ahead of print.