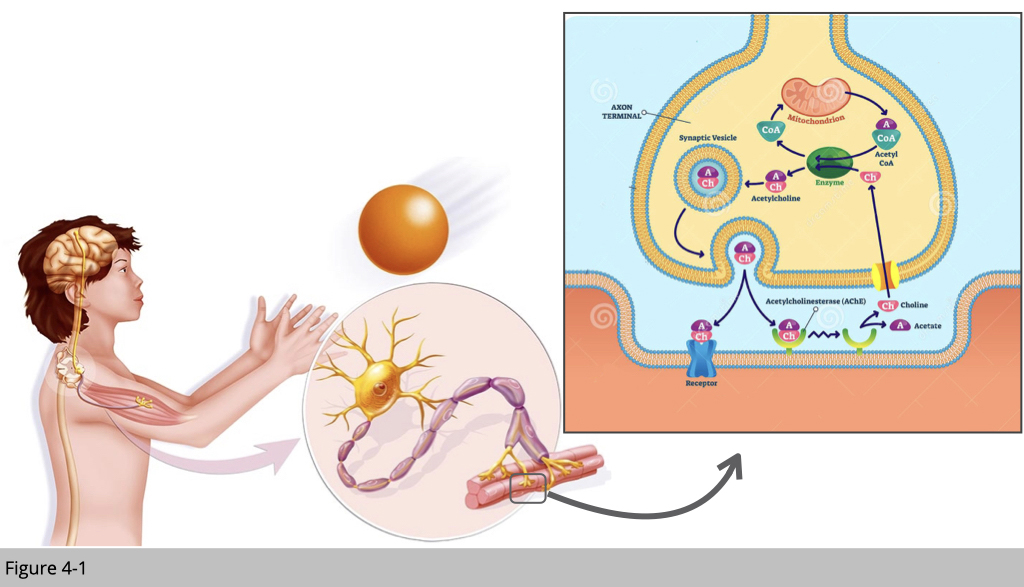
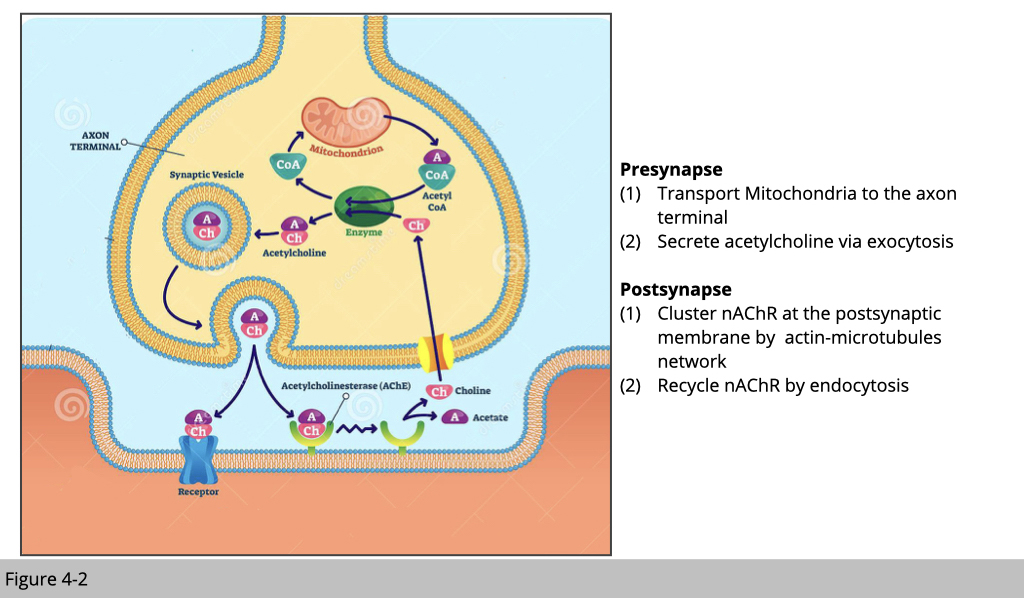
Figure 4-2 is a simplified cartoon of where motor neurons and skeletal muscle meet, which we refer it as a neuromuscular junction (NMJ). Acetylcholine is the chief neurotransmitter secreted from motor neurons to mediate skeletal muscle contraction. Acetylcholine is synthesized in the motor neuron by an enzyme, choline acetyltransferase, from the compounds choline and acetyl-CoA which are made in mitochondria. Acetylcholine will then be packed into synaptic vesicles and subsequently be released into the synaptic cleft.
Next, the acetylcholine molecules bind to the nicotinic acetylcholine receptor (nAchR) which is an ion-channel receptor on the muscle cell membrane, causing the sodium channels to open. Subsequently, sodium ions flow into the muscle cell, initiating a sequence of steps that finally produce muscle contraction.
What do those processes have to do with the cytoskeleton dynamic?
First of all, at the motor neuron presynapse site, we need a lot of good mitochondria to produce enough acetyl-CoA, so we can generate the acetylcholine molecules. In order to satisfy this high demand for acetyl-CoA, we need to make sure the fresh mitochondria can be consistently transported to the presynapse site, and the old/damaged one can be efficiently removed.
Second, the secretion of acetylcholine is via exocytosis which actin and myosin play an important role for secretory vesicles to their fusion sites at the plasma membrane and their release.
At skeletal muscle site- the postsynapse, in order to have synchronized muscle contraction triggering by a motor neuron, the nAChR needed to be clustered in the specific area, and this process of arrangement is dependent on both actin and microtubules network. Because of the high demand for nAChR at the postsynaptic site, our skeletal muscle actually recycle nAChR by endocytosis, which again actin and myosin mediate it.
Together in summary, the cytoskeleton dynamic is the fundamental mechanism controlling how we move.
Denervation causes nAChR loss, consequently induced muscle fibre loss.
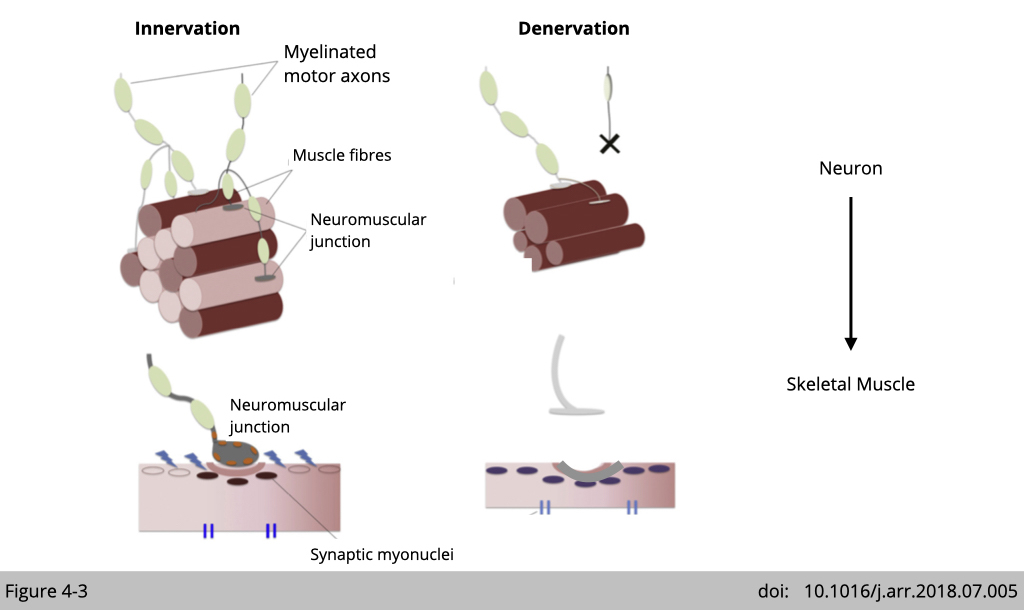
In order to maximize the force production and maintain the coordination in limb muscle, each lower motor neuron innervates a single muscle fiber, and each muscle fiber only gets innervated by one motor neuron. Mature nAChRs only clustered in the center of muscle fiber at where the motor neuron axon terminal is. Denervation of motor neurons in skeletal muscle causes axon degeneration and subsequently leads to muscle atrophy. From here, I think you will agree that motor neuron controls skeletal muscle movement and survive.
α-Bungarotoxin as a competitive antagonist to nicotinic acetylcholine receptors.
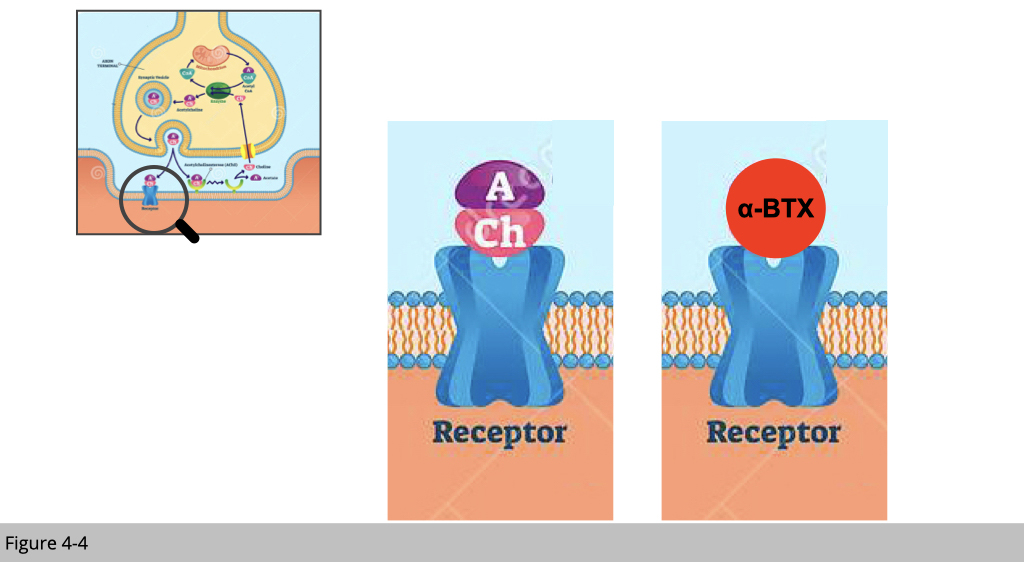
The connection of motor neurons and skeletal muscle is dynamically regulated. How could we see it?
We can study the dynamic of the neuromuscular junction (NMJ) by tracing the distribution of nicotinic acetylcholine receptor (nAChR) in the synaptic region. In this case, we use α-Bungarotoxin (α-BTX), which has high-affinity binding to nAChR. α-BTX is a 74-amino-acid α-neurotoxin extracted from the venom of the elapid Taiwanese banded krait snake. α-BTX can be conjugated to a fluorophore for immunofluorescence (IF) staining of fixed tissues and visualization via fluorescence microscopy. α-BTX has played a large role in determining many of the structural details of the nAChR.
Immunofluorescence staining of NMJ.
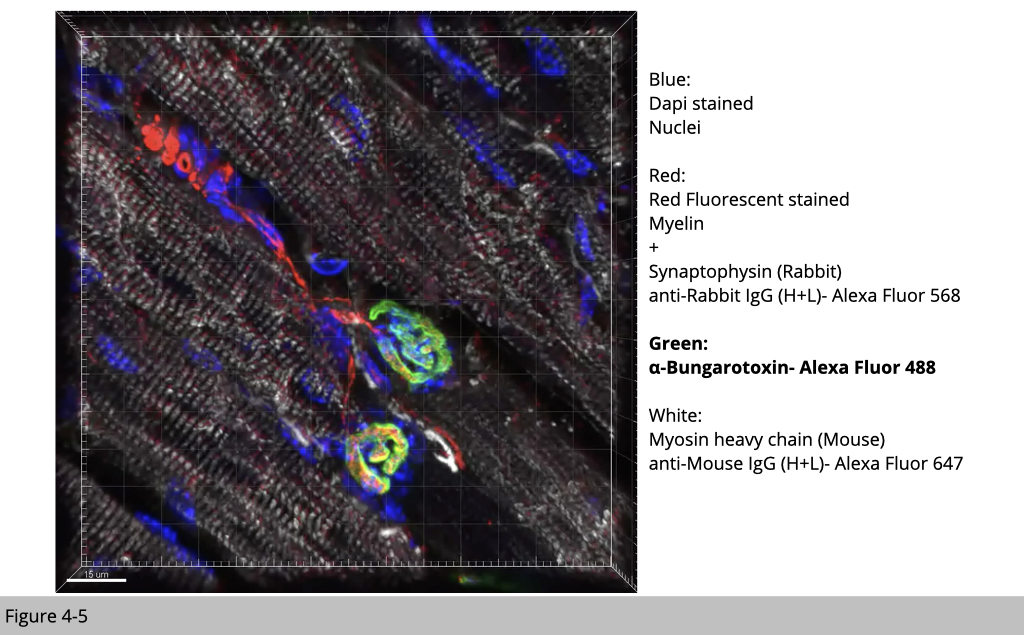
Figure 4-5 is an image of IF staining in mouse skeletal muscle. Applying IF staining of multiple antibodies and fluoroprobe to label motor neuron and skeletal muscle, you will see that each nAChR cluster labeled by α-BTX with green fluorescence is near to motor neuron labeled by myelin filament and synapse vesicle in red. The muscle fiber is labeled with muscle type myosin heavy chain in white, and nuclei were stained by DAPI, a fluorescent stain that binds strongly to adenine–thymine-rich regions in DNA, in blue.
Note that one postsynaptic nAChR cluster is conjugated with one motor neuron axon. While most of myonuclei is on the periphery of individual muscle fibers and separated from each other with almost perfect distance, there are a group of myonuclei accumulated underneath the site of nAChR cluster. These postsynaptic myonuclei are specialized to express genes required for NMJ structure and function.
Increased fragmentation of postsynaptic unit and decreased the connection to neuron (denervation) in aged NMJ.
During the aging process, we would walk slower and we would feel that we are much weaker than our 20’s or 30’s. Loss of muscle strength proceeds muscle atrophy. So what happens?
The simple answer is that we do not know. How come? You might ask. Muscle is not a difficult “organ” or tissue to study, right? It is not hidden under the skull like the brain or spine. We should easily get a biopsy to study its morphology and molecular changes over time in the model organisms or even humans.
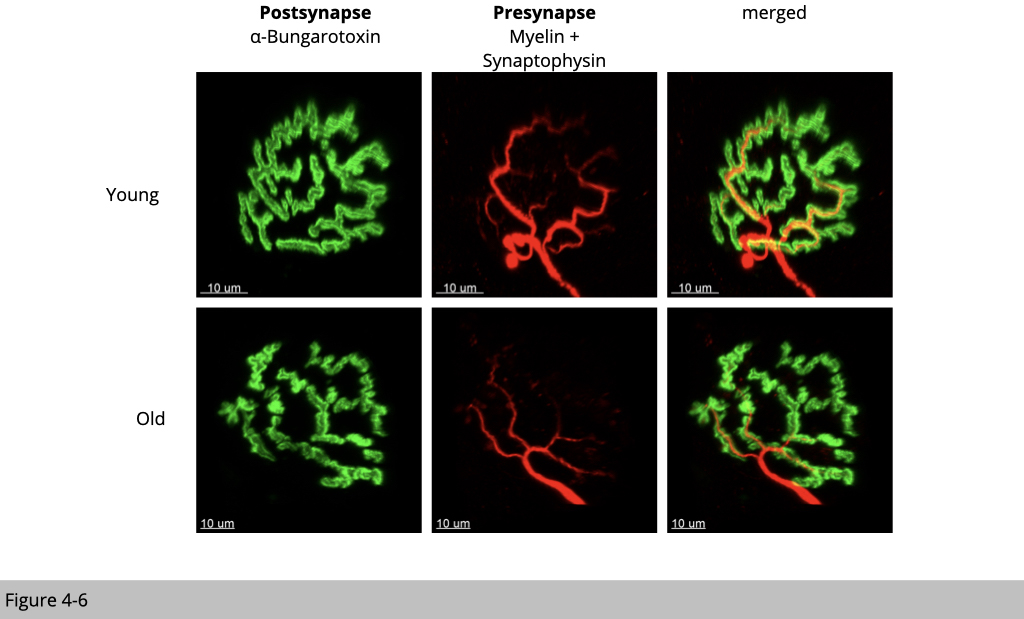
What we do know is that aging rodents such as rats and mice (the popular model organisms to study human disease) have fragmented nAChR clusters and those nAChR clusters are denervated. Figure 4-6 is the IF staining of NMJ structure from 6 months of age (young) and 26 months of age (old) male mouse skeletal muscle. You could see that the green signal from α-BTX labeling of nAChR is discontinued suggesting the fragmentation of nAChR clusters and red signaling from staining of motor neuron markers is reduced indicating denervation in old mouse muscle.
In contrast to rodents and other species, there is much less known about human NMJs at any stage of life. The limited studies carried out to date have revealed that, compared to rodents, human NMJs are significantly smaller and exhibit a simpler topology. The effect of aging on human NMJs is less clear. Moreover, most information documented in humans is “associated” studies in muscle abuse or exercise. When we don't use our muscles, both motor neuron, and skeletal muscle shrink. The obvious phenotype of muscle disuse is that the denervation occurred at the neuromuscular junction. In contrast, when we exercise, especially for resistance exercise, skeletal muscle grows in diameter and increases innervation.
It’s a chick and egg question. Does motor neuron guide the formation of NMJ or skeletal muscle attract motor neuron to initiate NMJ development?
Those of you who are believers of the soul theory~ that the brain makes up everything of our body. You probably will claim that it is neurons that have magic to make skeletal muscles move.
Postnatal synapse elimination is essential to achieve proper synaptic connectivity.
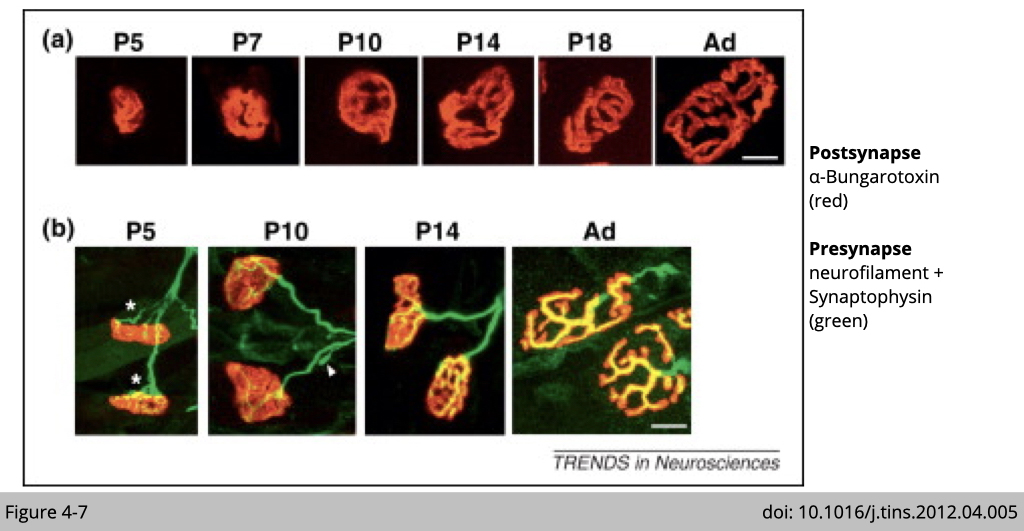
Yet, you are partially right. Figure 4-7 shows the NMJ formation during early postnatal development in the mouse. On panel a, what you can see that the size of the nAChR cluster is increased when the mouse gets matured (P indicates the postnatal day immediately after birth and Ad referes as adult).
Panel b shows that there is more than one motor neuron connected to one nAChR cluster/per fiber at the early stage of development. Yet, there is only one motor neuron connected to one nAChR cluster when the mouse gets matured. The elimination of axonal inputs occurs at the initial poly-innervated endplates (asterisks) during the first two postnatal weeks. It makes sense that only one motor neuron controls one muscle fiber. If multiple motor neurons are sending different signals to the muscle, it will generate confusion in the movement, at least in the limb muscle. (Postsynaptic AChR clusters and presynaptic nerve terminals are labeled by α-BTX (red) and synaptophysin/neurofilament (green), respectively. Scale bars, 10 mm.)
So, how does this motor neuron elimination occur?
Nerve-derived protein agrin activates muscle-specific kinase to cluster nAChRs through the synapse-specific cytoplasmic protein rapsyn.
The fundamental question is “how did nAchR clusters develop in the first place?”
We now know that all the mature nAchR clusters are located on the central region of skeletal muscle fiber (at least for limb muscle). It is like that a motor neuron has a ruler literately measuring every single muscle fiber and find the center to form their connection.
Wasn’t it amazing?
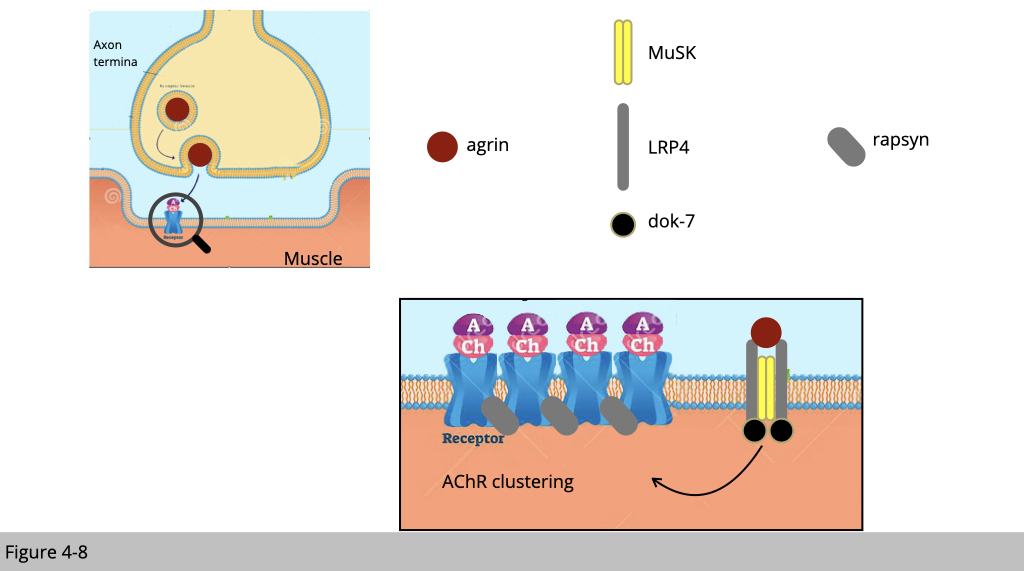
Since the motor neuron could order muscle contraction, it is believed that the signal for NMJ formation is initiated from the motor neuron. Motor neurons release the molecule agrin to bind LRP4, a member of the low-density lipoprotein receptor, in skeletal muscle. LRP4 will then activate muscle-specific kinase (MuSK) to trigger nAChR insertion to the nearby membrane. The full activation of MuSK also required its binding to Dok-7, a muscle adaptor protein.
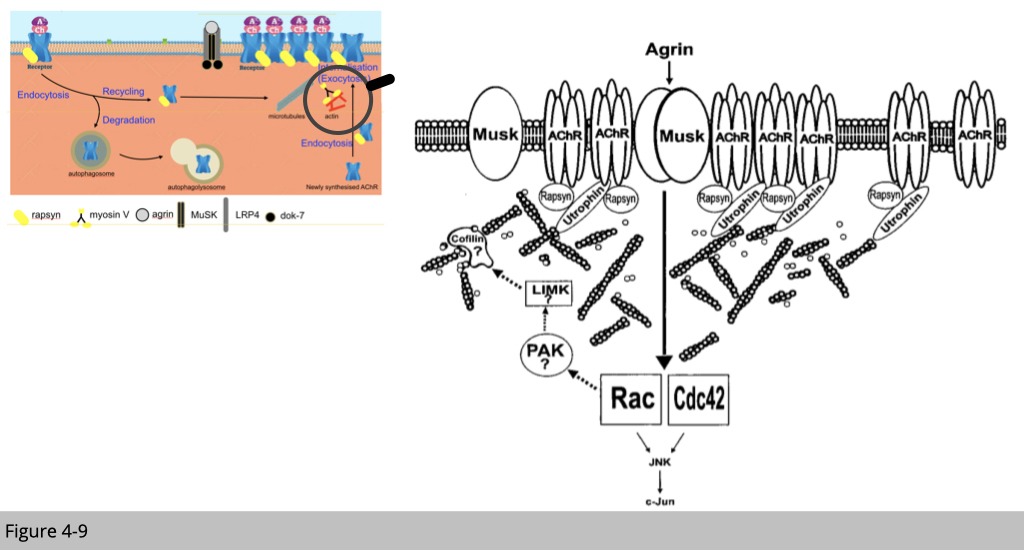
The full activation of MuSK leads to the reorganization of the nAChR-associated actin cytoskeleton through the modulation of Rho family GTPases and their downstream molecules. In addition, Rapsyn, a membrane-associated cytoplasmic protein that anchors nAChRs at the muscle membrane, is important for MuSK-induced nAChR clustering and stabilization.
Recently, additional functions of agrin have been discovered, including the up-regulation of gene transcription in myonuclei and the control of presynaptic differentiation. Agrin, therefore, appears to play a unique role in controlling synaptic differentiation on both sides of the NMJ. This simple cartoon showed in Figure 4-9 is a summary of multiple knockout mouse and human mutation studies.
Nerve-independent initiation of postsynaptic development.
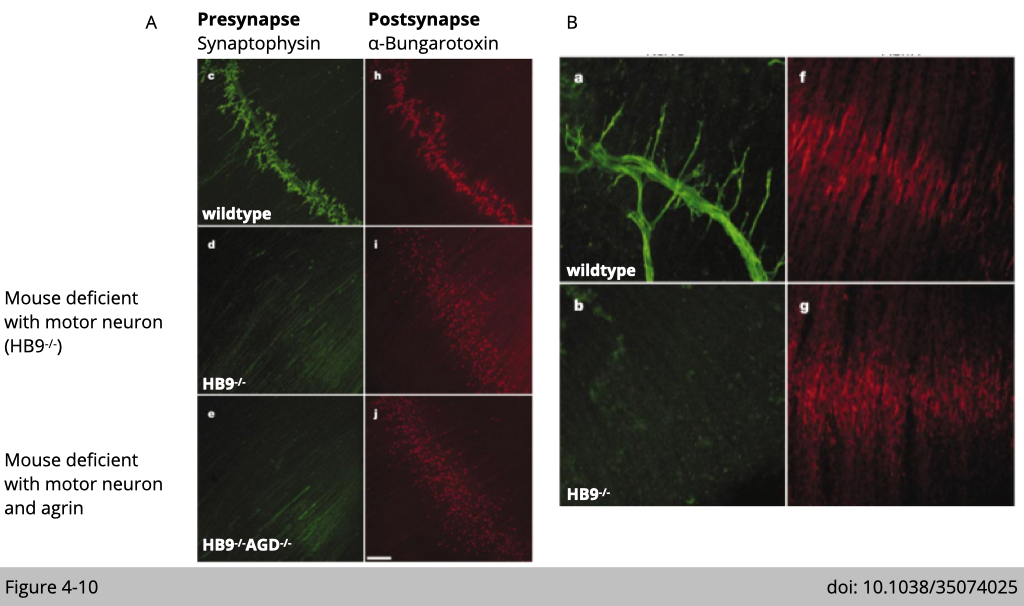
Kuo-Fen Lee’s group at Salk Institute generated multiple mouse models to find out which component initiates nAChR clustering in skeletal muscle. One is to knock out HB9, the motor neuron-specific transcription factor, to eliminate motor neuron formation, so the diaphragm muscle develops without innervation. The second transgenic mouse line is the double knockout of HB9 and agrin, a neurotransmitter that initiates LRP4-MUSK signaling in skeletal muscle. They examine the NMJ formation in the diaphragm muscles from E18.5 emberyo. In the Figure 4-10A shows that motor neuron pre-synapse terminal was labeled by antibody targeting synaptophysin, an integral membrane protein localized to synaptic vesicles, in green. nAchR was labeled by α-BTX, a neuron toxic specific targeting nAChR, in red. Surprisingly, both knockout mouse lines have the positive signaling of α-BTX labeled nAChR in skeletal muscle suggesting the formation of NMJ (Figure 4-10A, i and j).
The detection of nAChR clusters was not only present in mutant embryo diaphragms but also formed an end-plate band in the center of the muscle (Figure 4-10B). The distribution of nAChR clusters was similar in mutants and controls embryo muscle, although nAChR clusters from the mutant embryo were significantly smaller. Together, these results indicate that initiation of postsynaptic development is independent of nerve.
MuSK is required for initiation of synaptogenesis.
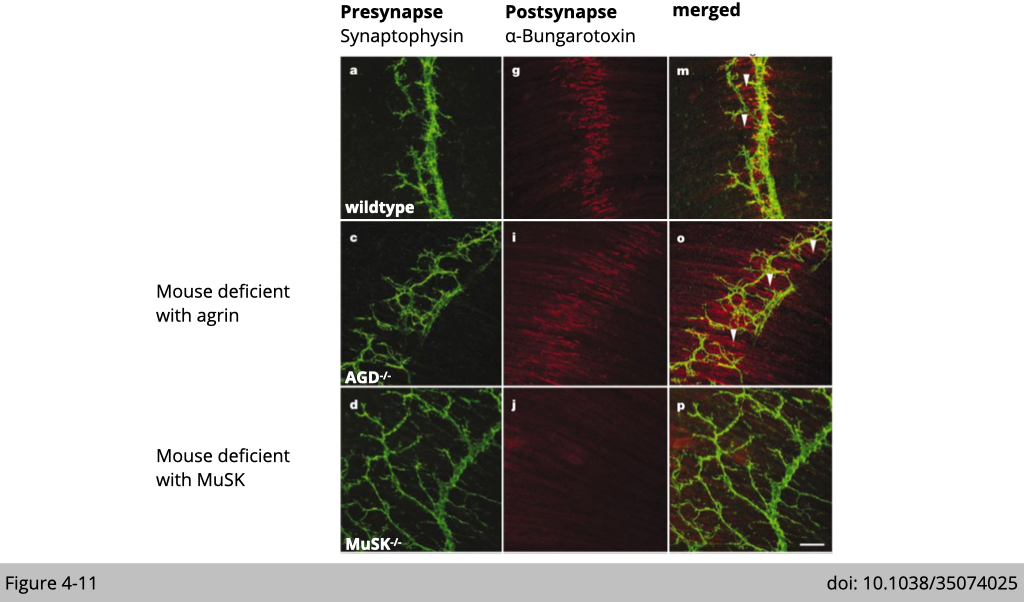
Since the motor neuron is not essential for initiation of nAChR clusters, next Lee’s group test whether the muscle is responsible for this process. They generate the third knockout mouse line deficient with muscle-specific kinase (MuSK). As expected, there is no nAChR cluster detectable throughout the developmental stages in the MuSK knockout embryo (Figure 4-11). These results suggest that initiation of postsynaptic differentiation is dependent on the signaling from the muscle.
This elegant piece of genetic work demonstrates that the formation of the nAChR cluster is pre-initiated in the skeletal muscle. Meaning that the preclustering of nAChR is independent of nerve signal but is more relied on signaling from the skeletal muscle.
Multiple steps in postsynaptic differentiation in the neuromuscular junction.
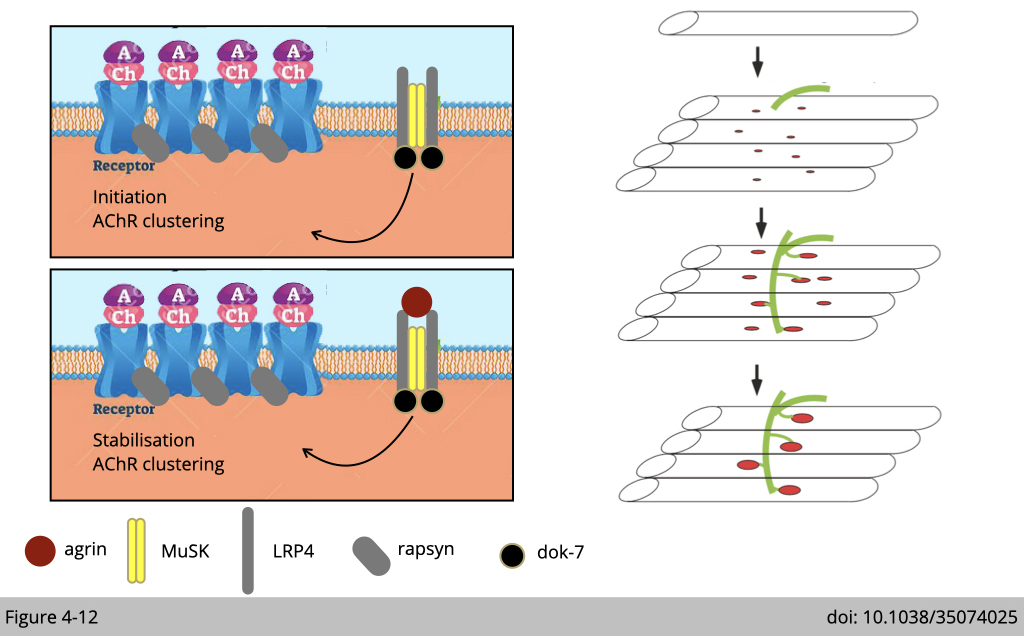
Even thorough the pre-clustering of nAChR mainly relies on the signaling from muscle, the maintenance and stabilization of the nAChR cluster are definitely required neuronal signals. Without agrin, the pre-clustering nAChR could not be maintained after birth and result in lethality.
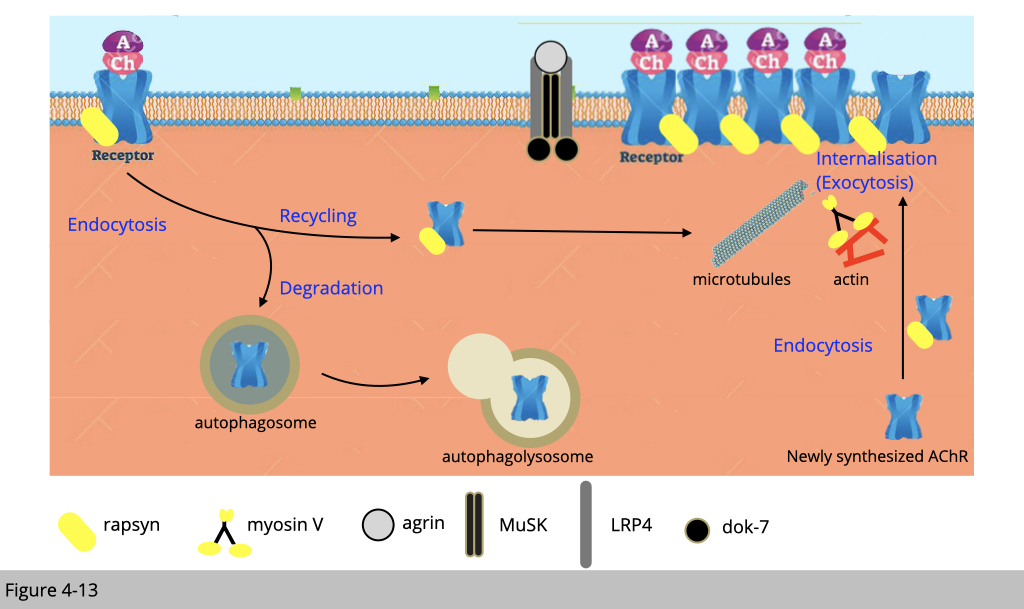
A high density of nAChRs at synapses is required to initiate a synaptic action potential in the myofiber. Newly synthesized nAChR was assembled in the endoplasmic reticulum from individual subunits (α, β, γ/ε, δ) and internalized to the membrane through exocytosis via myosin V, our swing guy.
At fully functional and mature synapses, nAChRs have a half-life of about 14 days in adult mice. nAChRs are degraded via macroautophagy/autophagy. Yet, with the high demand for nAChR for muscle contraction, a significant portion of nAChR is recycled to meet the demand. The recycled nAChRs turn over more rapidly than pre-existing or newly synthesized nAChRs that are colocalized in the same postsynaptic membrane.
How to study it?
Streptvidin-Biotin complex method for immunostaining.
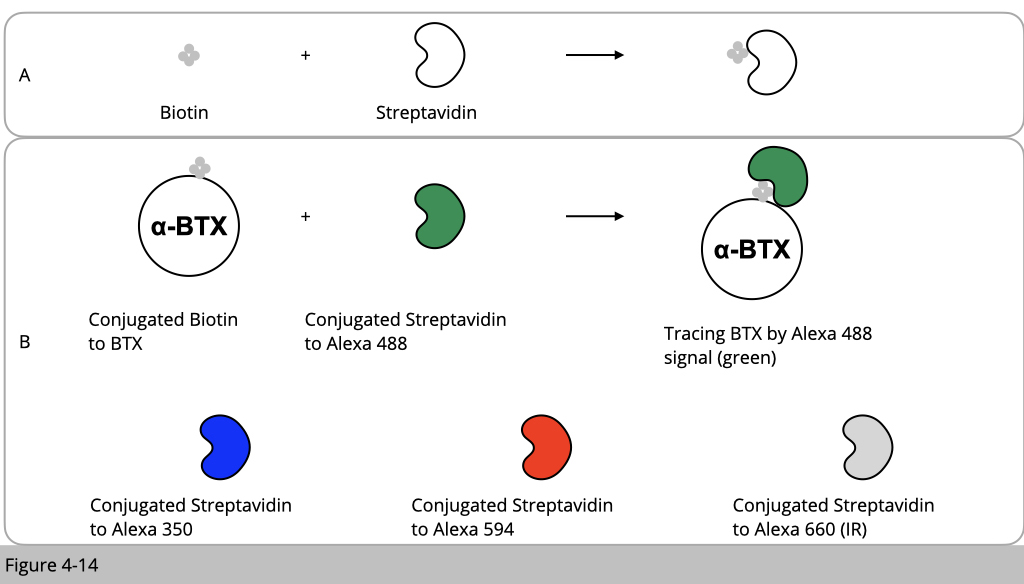
The way to study the stability of nAChR is brilliant. It might seem complicated initially. I hope that you will appreciate the creativity, whoever the first person designs this type of experiment in the end of this part.
We know that α-BTX has a very high affinity and selectivity to bind nAChR. α-BTX is basically a small chemical molecular which can be synthesized and conjugated with any molecular such as different color of fluoroprobes. For example, we can conjugate α-BTX with rhodamin fluoresce in red. Then all the nAChR will be labeled in red. Therefore, α-BTX is ideal chemical tool to study the stability of nAChR.
One more concept I need to introduce to you. It is about biotin and streptavidin. The interaction between streptavidin and biotin is one of the strongest non-covalent interactions in nature (Figure 4-14A). The streptavidin-biotin system is a widely used tool to study protein-ligand interactions. Using this trick, we can then conjugated α-BTX with biotin and conjugated streptavidin with different fluoroprobes which have a different color (Figure 4-14B).
By selectively labeling different nAChR pools (preexisting, recycled, and new) within a single synapse in the living mouse, we were able to estimate the contribution of each pool to the NMJ over time.
Labelling of distinct nAChR pool with different conjugated α-BTX.
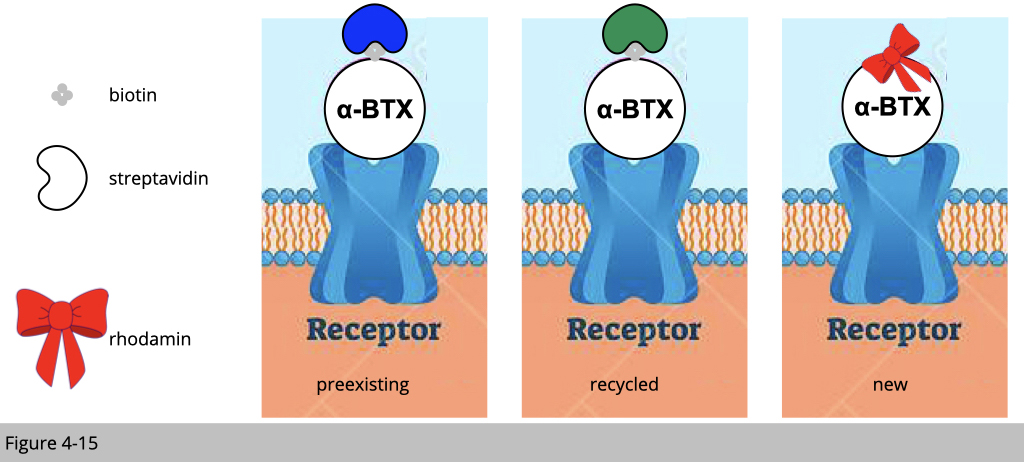
Figure 4-15 is the summarize of the experimental design. We are going to label pre-existing nAChR with biotin-conjugated α-BTX plus blue streptavidin and the recycled nAChR with biotin-conjugated α-BTX plus green streptavidin. The newly synthesized nAChR will be labeled with rhodamine conjugated α-BTX in red. How could we do it?
In vivo imagine of neuromuscular junction.
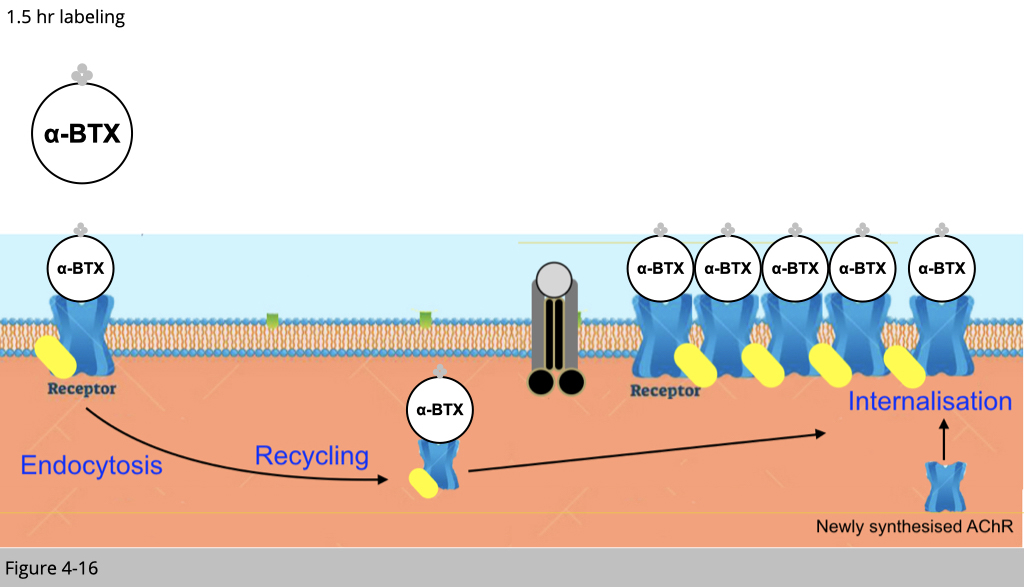
First of all, we inject mice with one shot of overdosed biotinylated-αBTX (no color). By doing so, the preexisting nAChR and the nAChR undergoing recycling would be bound with this biotinylated-αBTX.
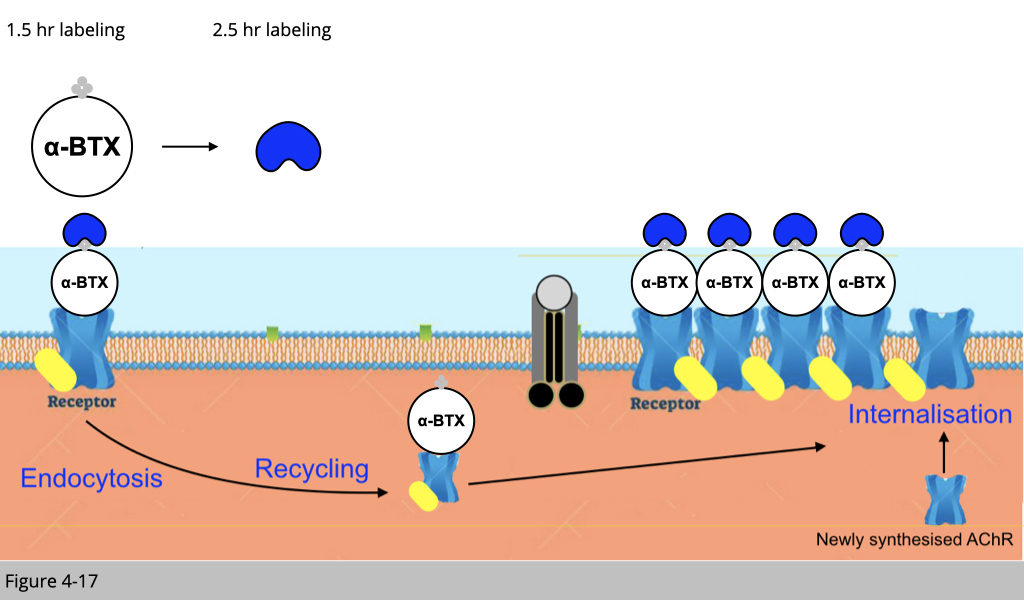
After 1.5 hrs, we then inject mice with one shot of blue streptavidin. Rule number 1, all the binding of the small molecular we are using here would only occur on the plasma membrane of muscle fiber. Therefore, the blue streptavidin can only bind the preexisting nAChR labeled with biotinylated-αBTX on the surface of the plasma membrane.
The recycled nAChR which was also labeled with biotinylated-αBTX 1.5 hr ago would not bind to the blue streptavidin since the recycled nAChR is now in the cytoplasm.
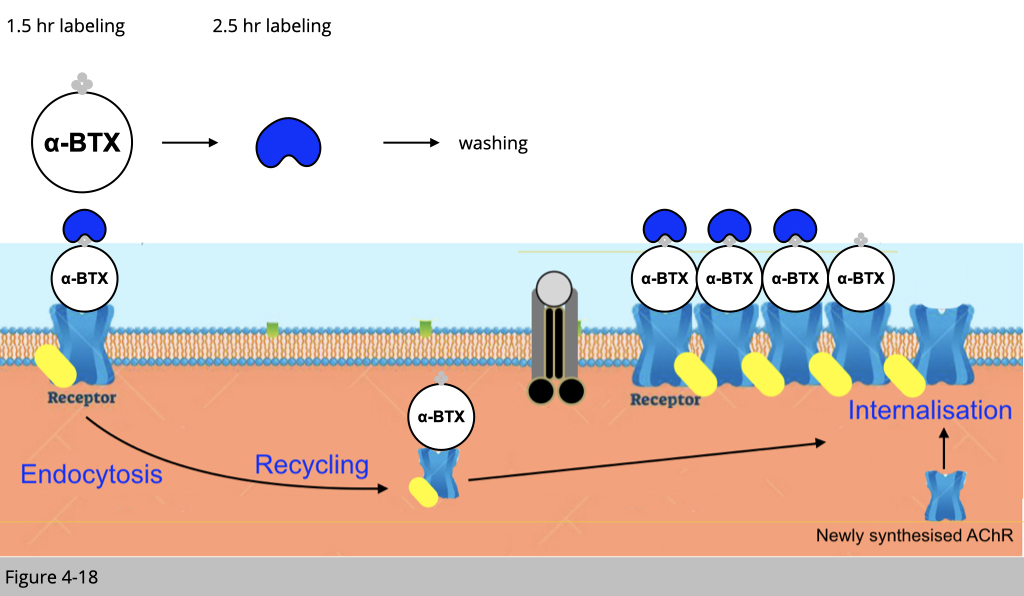
After 2.5 hr injection of blue streptavidin, we then wash any unbind blue streptavidin. The recycled nAChRs, which also bind to biotinylated-αBTX four hours ago, should be able to be internalized to the plasma membrane.
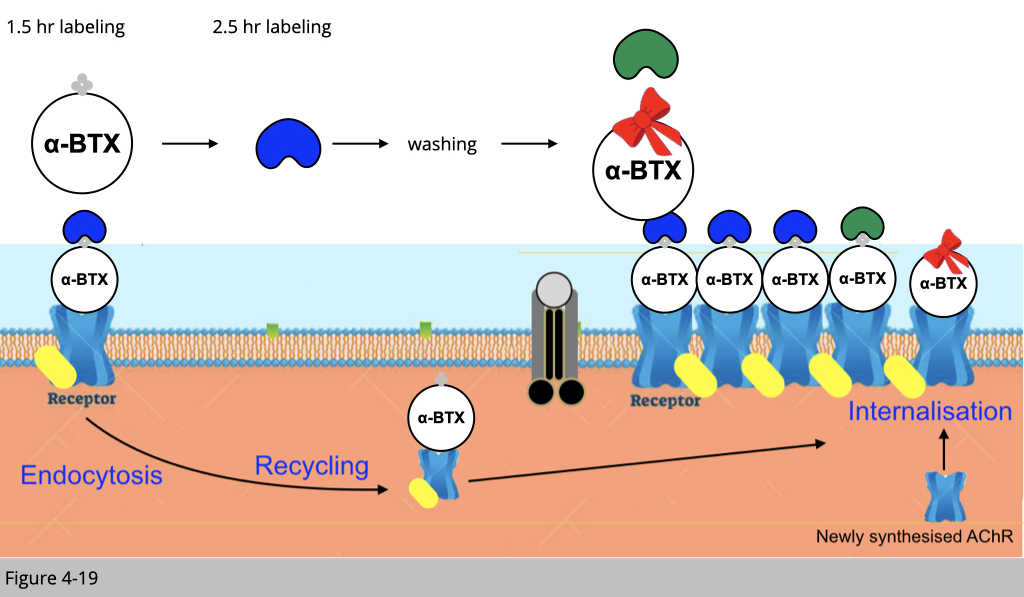
Finally, we inject mice with green streptavidin to bind the recycled biotinylated-αBTX labeled nAChR and fresh rhodamine conjugated αBTX to label newly synthesized nAChR. Under confocoal microscope, we can then reconstruct the 3D image of a single NMJ to differentiate the pre-existing nAChR labeling in blue, the recycled nAChR labeled in green and the newly synthesized nAChR labeled in red.
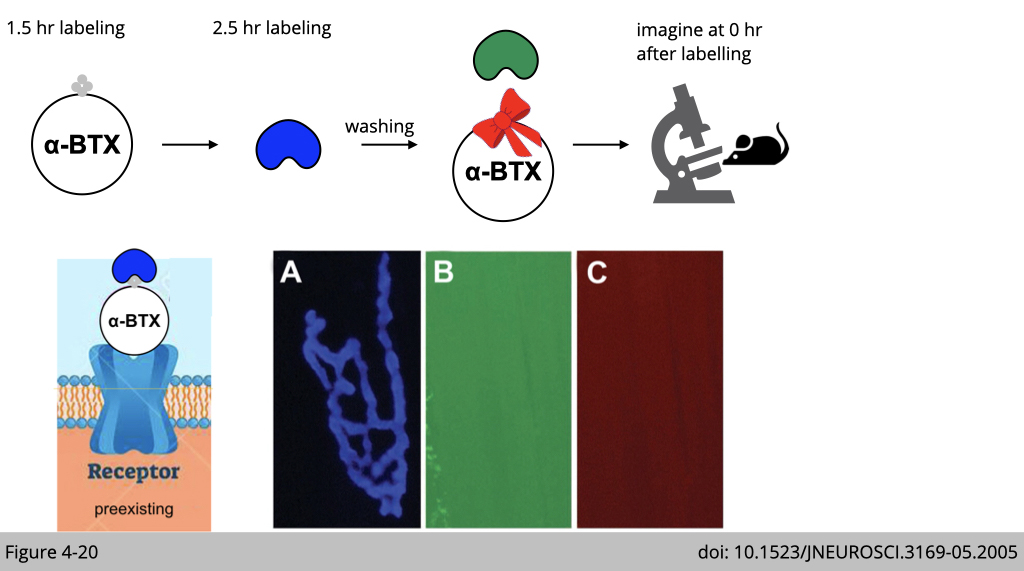
Figure 4-20 shows the labeling result imaged from a living adult mouse. A selective NMJ was imaged and labeled with a saturating dose of biotinylated-αBTX followed by a saturating dose of blue streptavidin. The absence of any labeling of green or red signal (even when camera exposure was increased significantly) indicates that all receptors and biotin sites were saturated by the initial applications.
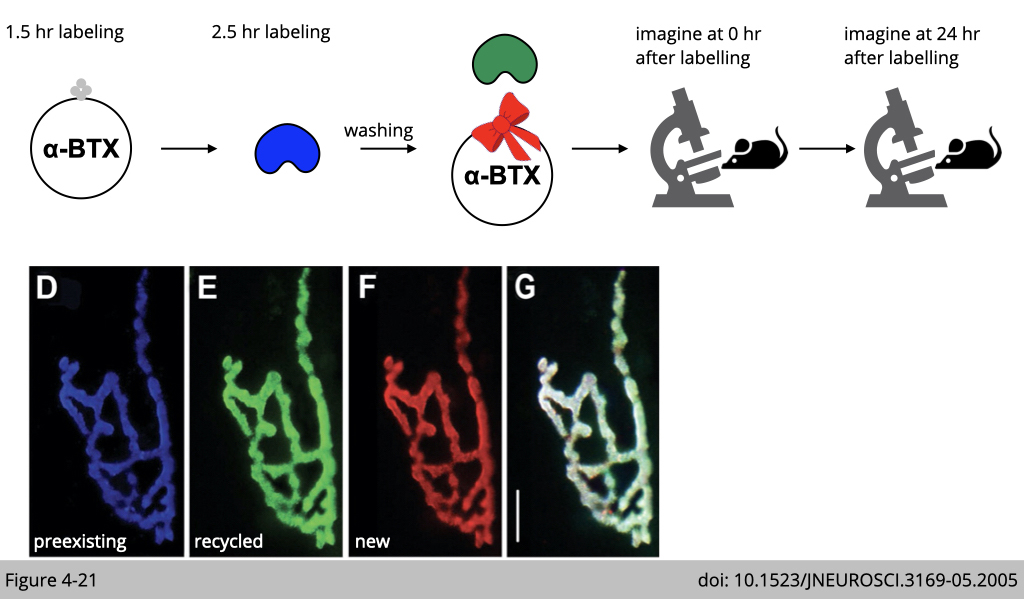
When the same synapse was imaged 24 h later (Figure 4-21), we can find that much of the preexisting nAChR labeled with blue αBTX remained, but there was also significant fluorescence on the green αBTX (labeled recycled nAChR) and red αBTX (labeled the newly synthesized nAChR).
Recycled nAChRs are removed more quickly than pre-existing nAChRs.
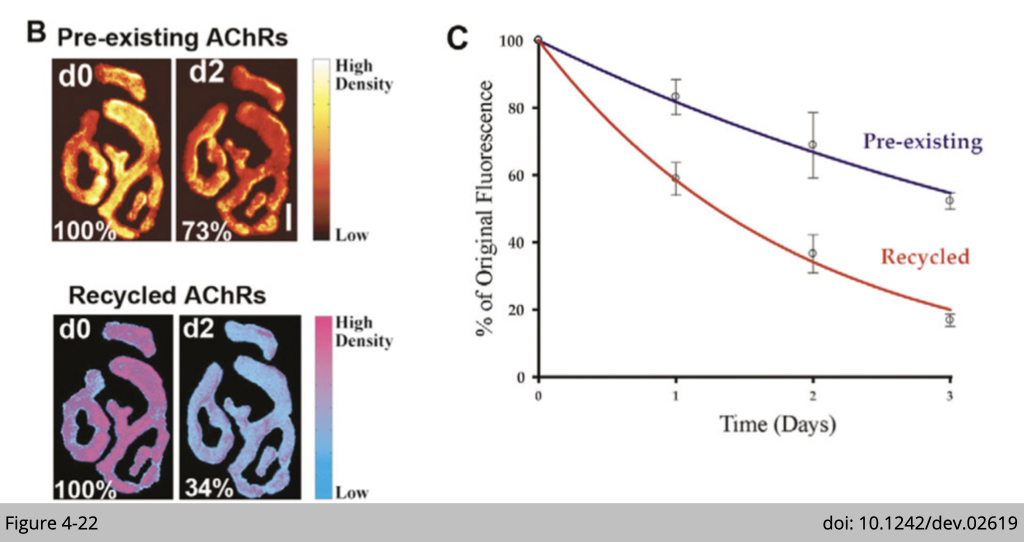
Using the technique described above, Mohammed Akaaboune's group at University of Michigan demonstrate that recycled receptors in functional synapses are removed approximately four times faster than pre-existing receptors (Figure 4-22). The sternomastoid muscle was labeled first with BTX-biotin/strept-488, then 3-4 days later with strept-594 to selectively label recycled AChRs. Preexisting and recycled AChRs labeled with strept-488 and strept-594 (Figure 4-22B) and assayed for fluorescence intensities immediately after strept labeling (d0), and 2 days later (d2). The total fluorescence intensity of each AChR pool was normalized to 100% at initial imaging. Pseudo-color images provide a linear representation of the density of AChRs (Figure 4-22C).
That’s how we measure the half-life of preexisting and recycled nAChR showed in Figure 4-22. Wasn’t it amazing? The same technique has been applied to different physiological settings such as denervation, muscle wasting, and aging to analyze the nAChR turnover rate.