Structural study: Using cyroEM to study motors.
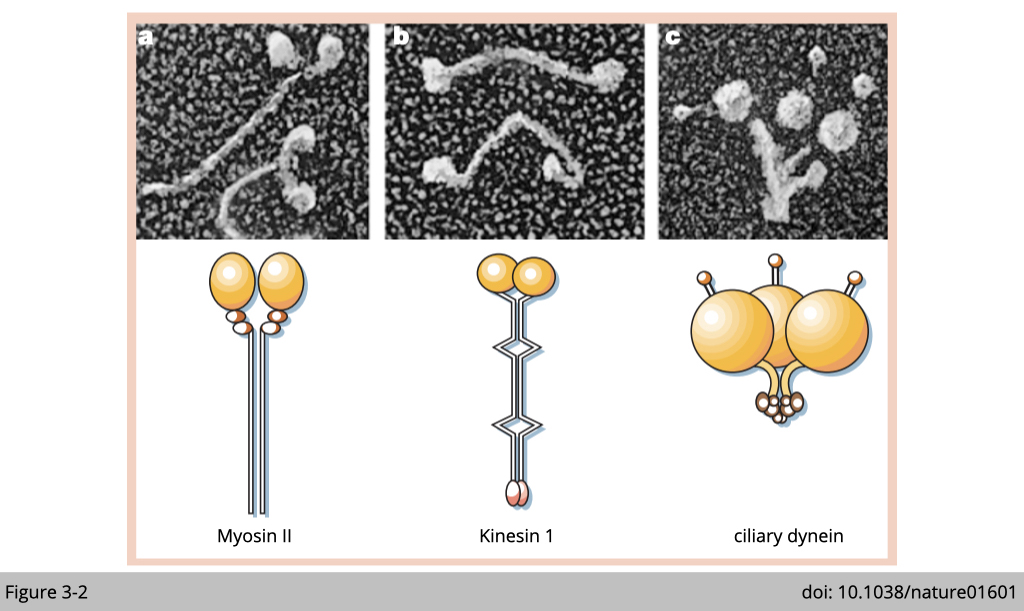
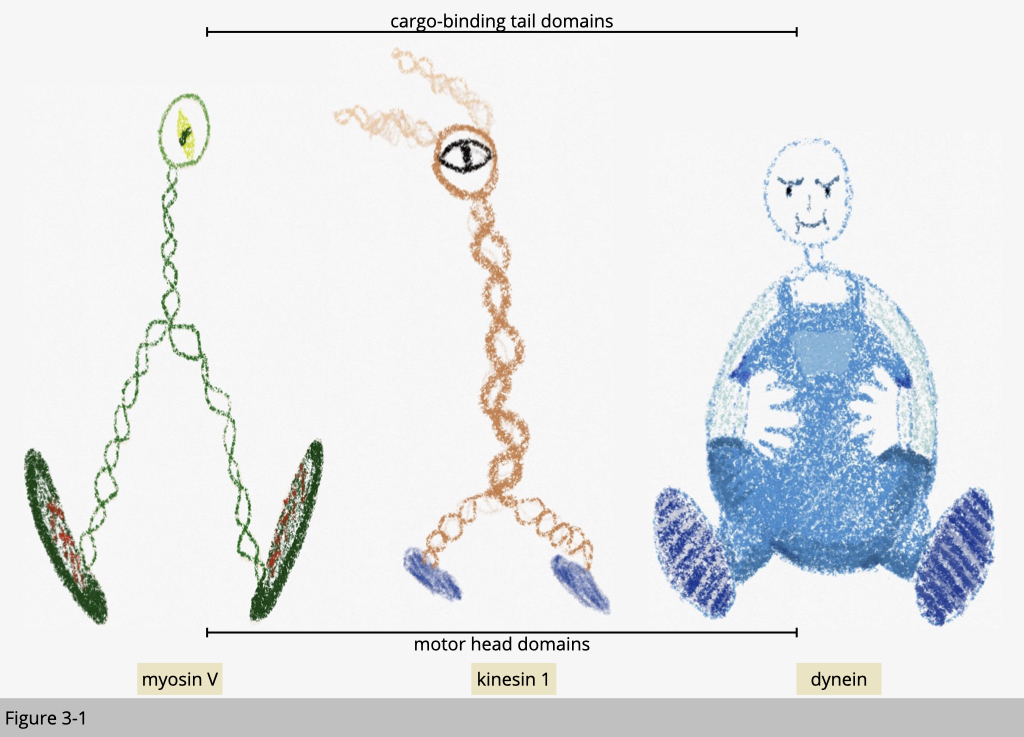
To begin the study of motor proteins, we could start with imaging their structure. For a motor to move on the ground, it should have a wheel-like structure for walking and at least two of them. Under EM, you could find a common feature of these three types of motor proteins. They all have a globular motor domain. These three motor proteins use their big head (colored in yellow in Figure 3-2) to interact and walk on the cytoskeleton track.
More readings: Crystal structure of protein
Genetic study: Known genetic abnormalities of myosins in human or mouse models.
Next, you will want to know how many of those molecular motors in our body, do they move differently, or do they transport different cargo, right?
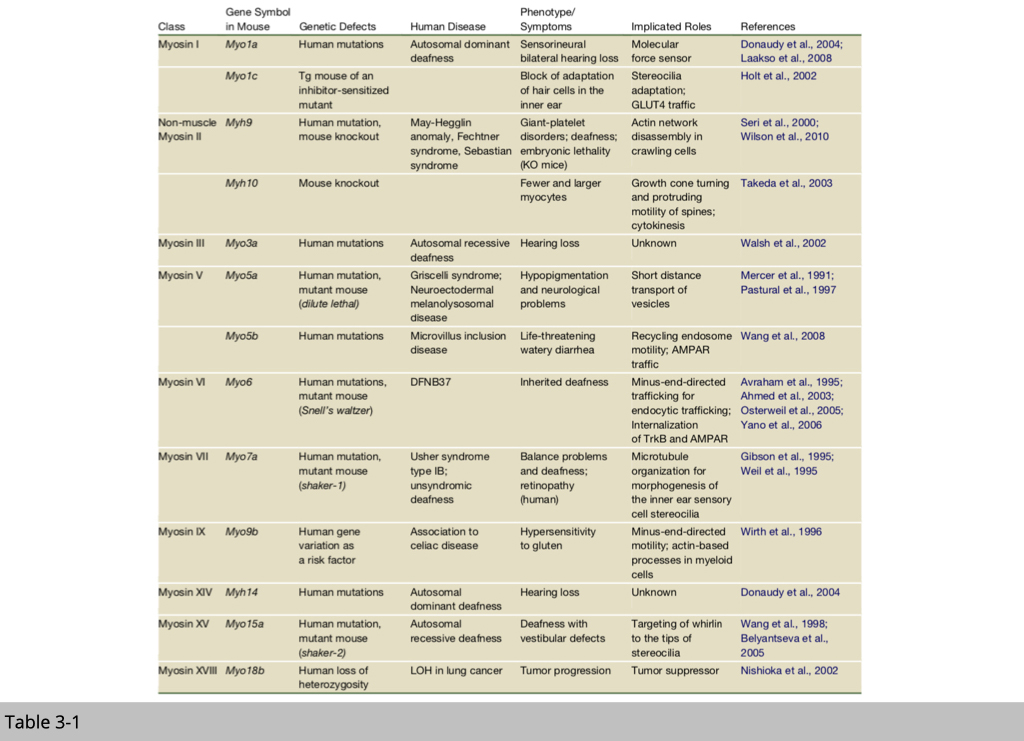
At this point, we know that mammals have genes for 40 myosins, over 40 kinesins, and more than a dozen dyneins. Many motors are not yet characterized, and clear functions are assigned to only a small subset. So far, they are quite a few mutations have been identified and linked to human diseases. Several transgenic /knockout mice have also been generated to study those motors’ functions.
Note that, the functions listed in table 3-1, most of them are not critical for survival. It does not mean that those myosins are not important for living. If the mutation or knockout of certain myosin is embryonic lethal, we won't have an alive human or animal to study.
Mechanical Data: Monitoring movements mediated by kinesin
Now we know how those motor proteins look like, how many of them, and which mutations cause diseases related to cell movement and cargo transportation. The final question is that how exactly they walk on the cytoskeleton?
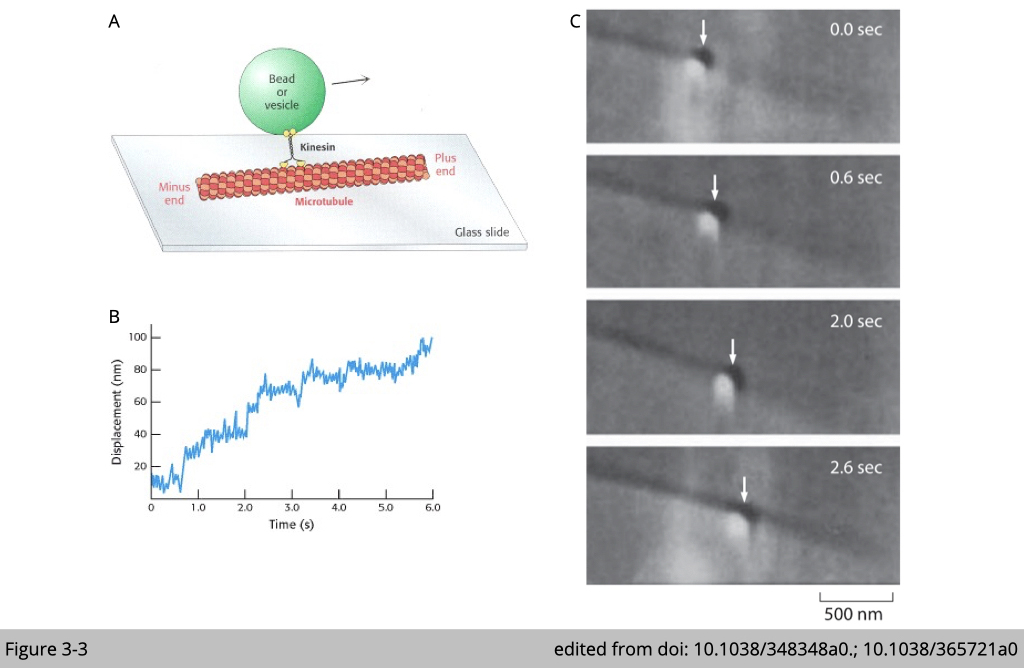
In Figure 3-3A, the drawing demonstrates a classical in vitro experiment to visualize how motor proteins move on the cytoskeleton. Firstly, a single kinesin molecule was incubated with glass beads for visualization. Then the complex is loaded into contact with a microtubule that was bound to a glass surface. The movement of kinesin was traced by the movement of glass beads under microscopy. By this in vitro setting, we can visualize the pattern of their movement (Figure 3-3C) and measure the speed of each motor protein (Figure 3-3B).
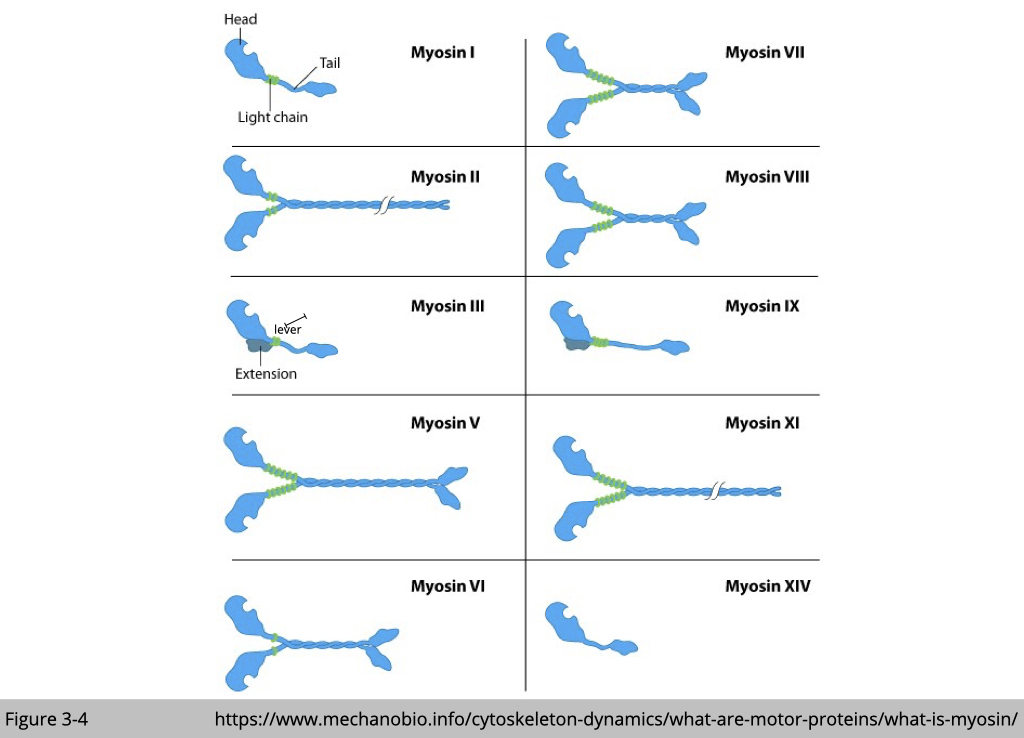
For myosin, there are 35 classes of myosin motors, made by at least 40 different genes. All of them have actin- and ATP-binding sites in their conserved catalytic head domains. The globular head of myosin binds actin in the presence of ADP. Replacement of ADP by ATP releases myosin binding to actin. After the hydrolysis of ATP, the energetic myosin heads change their angle toward a new front position and re-load on the actin filament, which in turn causes the movement.
Each myosin is somehow moving differently and takes a different size of the step. For example, myosin II only takes a step that is 7 nm long while myosin V could take a step up to 36 nm. That’s because the size of the step is limited by the length of the lever. Myosin V lever is three times larger than myosin II.
The cross-bridge muscle contraction cycle.
Of these, the most studied myosin is the muscle type class II myosin. It was first discovered by Willy Kuhne from muscle extracts in 1864.
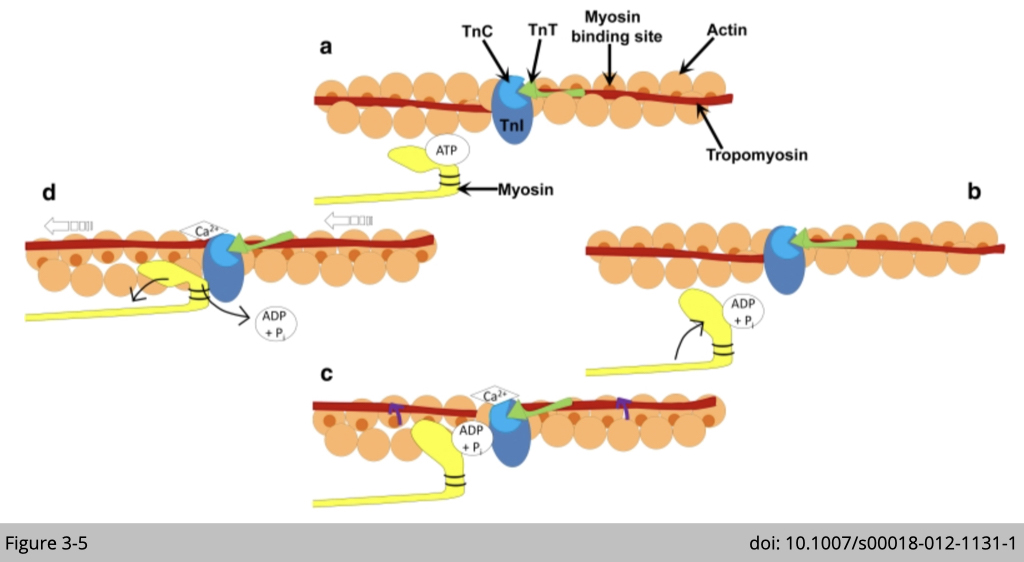
Muscle type class II myosins are found in skeletal and cardiac muscle, powering muscle contraction. The interaction between actin and type II myosin generates the force of movement by the formation of a cross-bridge. The globular heads of myosin bind actin, forming cross-bridges between the thick and thin filaments. The myosin heads then bind and hydrolyze ATP, which provides the energy to drive filament sliding. The speed of myosin II strides 6,000 nm/s in fast-twitch muscle.
Noted that, the interaction between skeletal muscle myosin II and actin is not only dependent on the energy state of myosin but is also regulated by calcium. Calcium binds to troponin (TnC) which induces the movement of tropomyosin on actin filament and strengths the binding between myosin and actin (Figure 3-5c). Therefore, the rate of ATPase and calcium transition (cytosolic calcium uptake and removal) are the major determinants of muscle contraction. We will discuss this subject in great detail in the later section about muscle.
Myosin motors are critical for neuroplasticity.
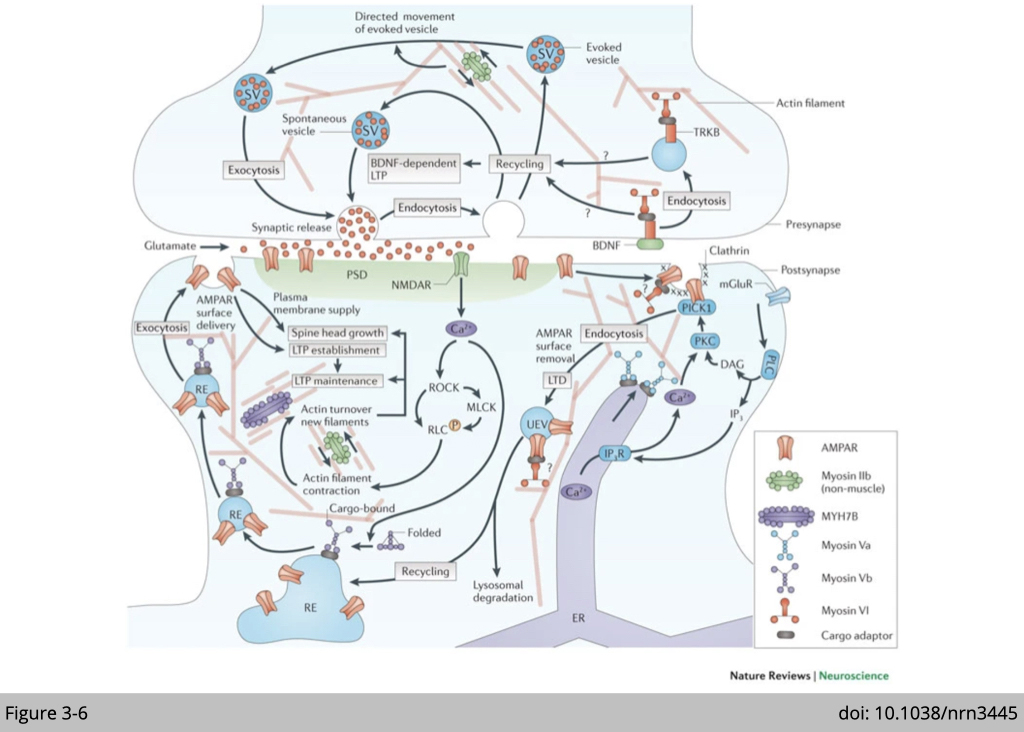
Besides force generation, the role of myosin in remodeling the network of actin filaments and cargo transportation has drawn tremendous attention in the study of neuroplasticity.
Presynaptic functions of myosins include: promotion of directed movement of synaptic vesicles (SVs) generated by endocytosis after evoked glutamate release by myosin II; and promotion of synaptic recycling of vesicles and induction of brain-derived neurotrophic factor (BDNF)-dependent long-term potentiation (LTP) by myosin VI. Postsynaptically, non-muscle myosin IIb and myosin Vb are activated by an NMDA receptor (NMDAR)-dependent calcium influx. Non-muscle myosin IIb promotes the turnover of actin filaments in spines, thereby contributing to spine head growth and the maintenance of LTP. Myosin Vb trafficks AMPA receptor (AMPAR) subunit GluA1-carrying recycling endosomes (REs) into spines and thereby contributes to spine head growth and AMPAR surface delivery during LTP establishment.
More readings: Neuroplasticity with Myosin Motors
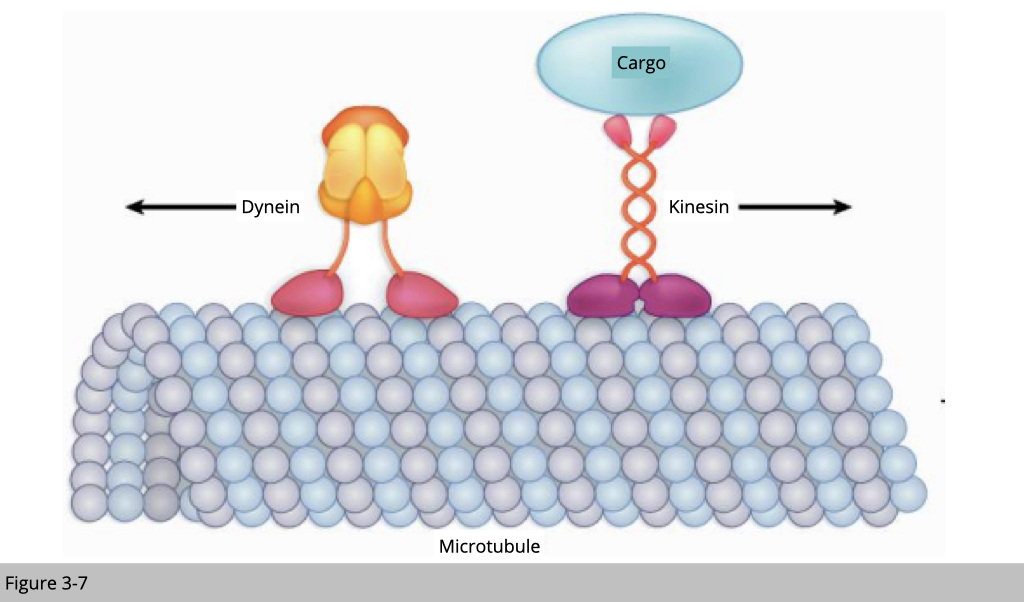
Kinesin moves along on the microtubule from minus to plus end, and dynein moves in the opposite direction. Let’s use an example of cargo transportation in the motor neuron. Kinesin carries newly synthesized mitochondrial from the cell body to the synapse. Dynein, on the other hand, moves back the old mitochondria in the autophagosome from synapse to cell body for degradation.
Kinesin walks forward on the microtubule like a soldier, step by step. The average step of kinesin is 8.6 nm long. The average speed of kinesin in anterograde axonal transport is around 1,800 nm/s. On the other hand, dynein walks like a drunken sailor. Dynein can walk around the microtubule, either right or left, back or forward, and take big or little steps. For its free mind, dynein walk with a bigger step around 20 nm compared to kinesin is only 8.6 nm and may be able to step around any molecular obstacle on the microtubule. The average speed of dynein in retrograde axonal transport is only around 1,000 nm/s, part of the reason is because of the way its walks.
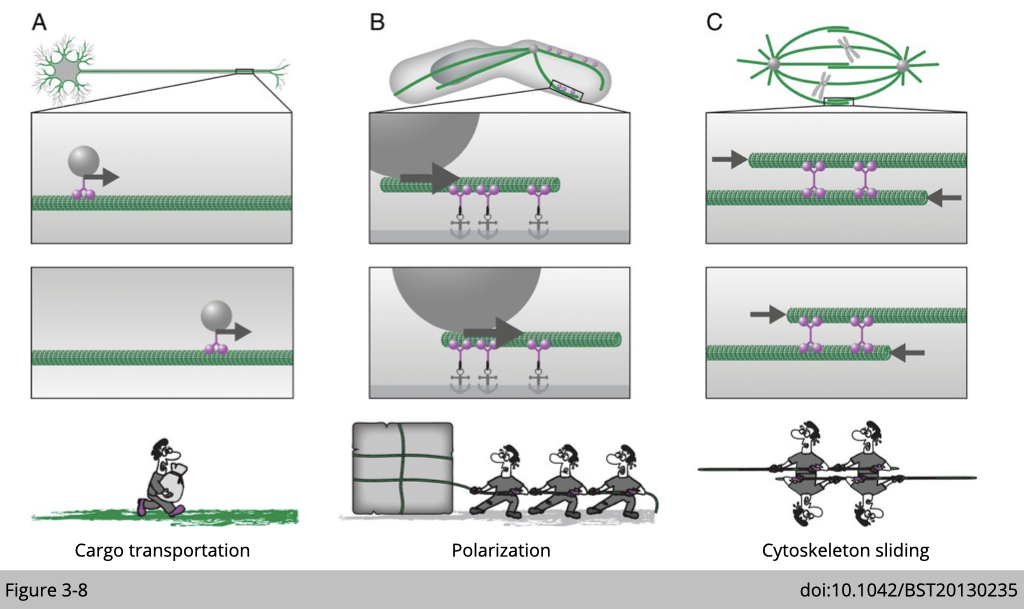
The fundamental difference between motor proteins and cytoskeleton-associated proteins is the ability of a motor protein to move along the cytoskeleton. Moreover, motor proteins use ATP hydrolysis to fuel their movement along the cytoskeleton within the cell. Therefore, they participate on
- cargo transportation
- set the polarization of cell, because each of them has their direction of moving
- cytoskeleton sliding, such as mitosis and skeletal muscle contraction
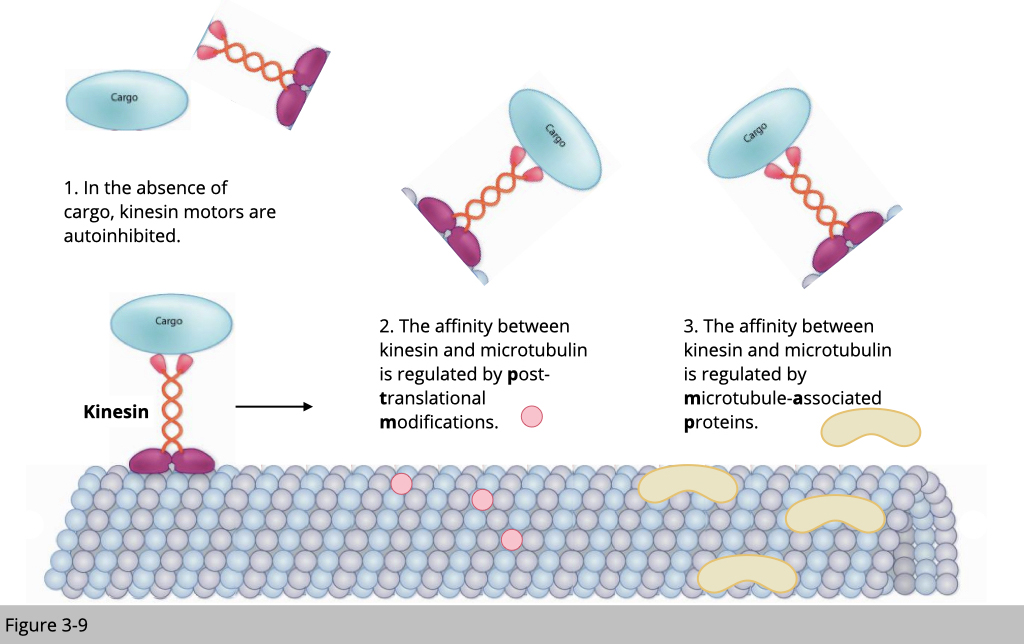
In this section, we will use kinesin-mediated transportation on microtubulin as an example to demonstrate how cells manage the traffic.
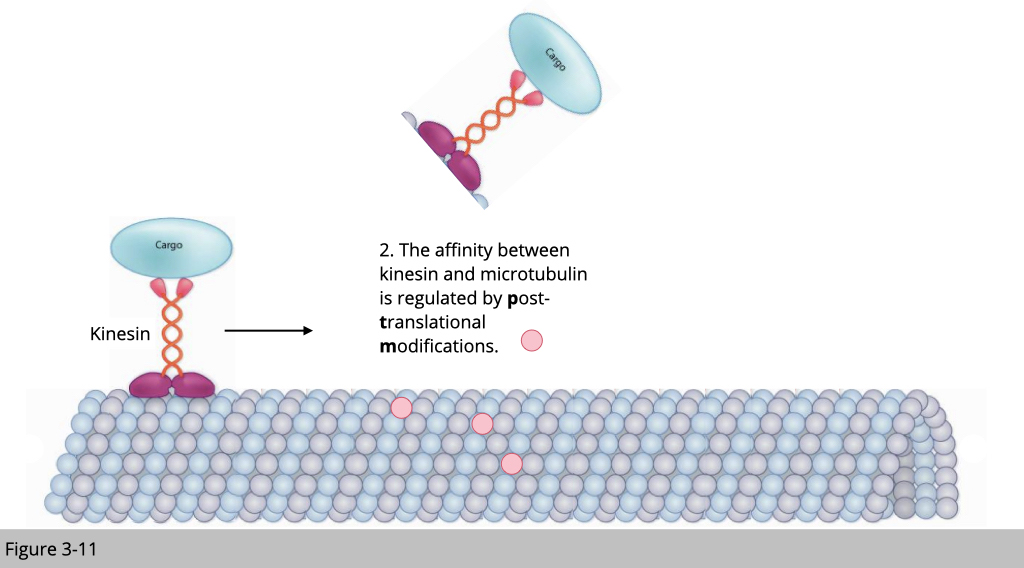
There are a couple of standard traffic rules for transportation. First, a motor protein without a cargo will be unloaded from the cytoskeleton track. This will prevent the non-used motor to jam the road. Second, the post-modification of the cytoskeleton could affect the structure stability and the arrangement of the cytoskeleton, which affects the speed of motor traveling. Third, the cytoskeleton-associated protein will also affect the recruitment and the unloading of the motor protein.
Post translational modifications of the motors
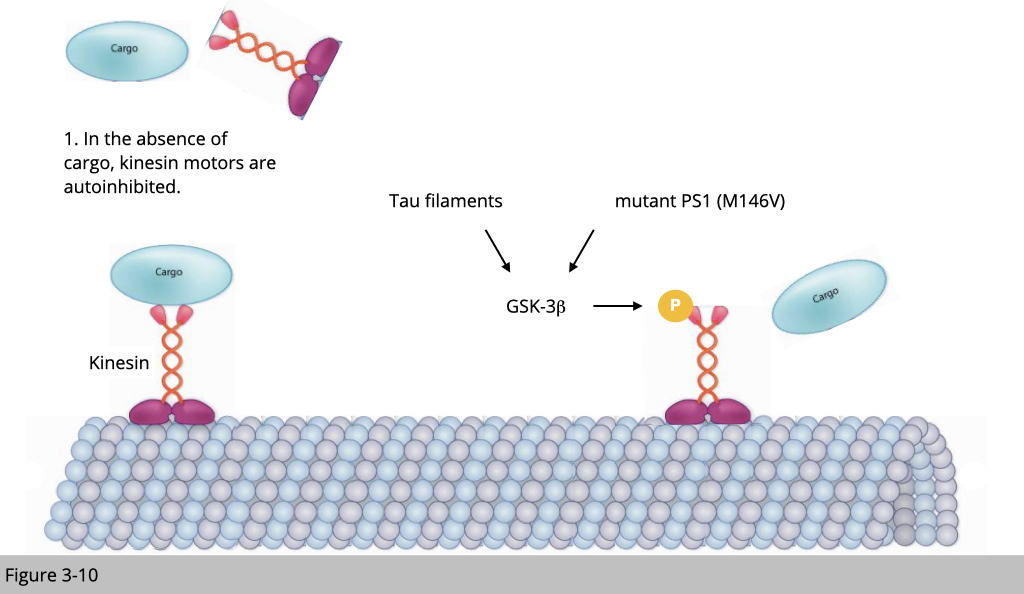
Hyper-activation of Glycogen synthase kinase-3 (GSK3) has been implicated in a wide range of neurodegenerative diseases. One of the mechanisms that underline GSK3 mediated neuronal pathology is to disrupt anterograde fast axonal transport. Phosphorylation of kinesin by GSK-3 causes the conformation changes of kinesin and forces it to unload its cargo. Tau filaments and mutant presenilin 1 (two known AD risk factors) activate GSK3β, therefore inhibit kinesin-dependent motility.
Post translational modifications of the track
Most of the studied post-translational modifications (PTM) of tubulin are found within the C-terminal tails of α- and/or β-tubulin. These tails are exposed to the outer surface of assembled microtubules and are key interaction sites for microtubule-associated protein (MAP). It is thus obvious that these PTMs of tubulin can influence, potentially in a selective manner, the interactions between microtubules and MAPs.
For example, detyrosination prevents the kinesin-13 unload from disassembling microtubules, thus providing a mechanism by which detyrosination could regulate microtubules stability. Another example of PTM is polyglutamylation. Polyglutamylation affects the structure and increases the negative charge of the intrinsically disordered tubulin tails, which are important for microtubules interactions with a broad variety of MAPs. Excessive polyglutamylation of microtubules has been shown to impair mitochondria transportation and cause axon degradation.
Microtubule-associated proteins (MAPs)
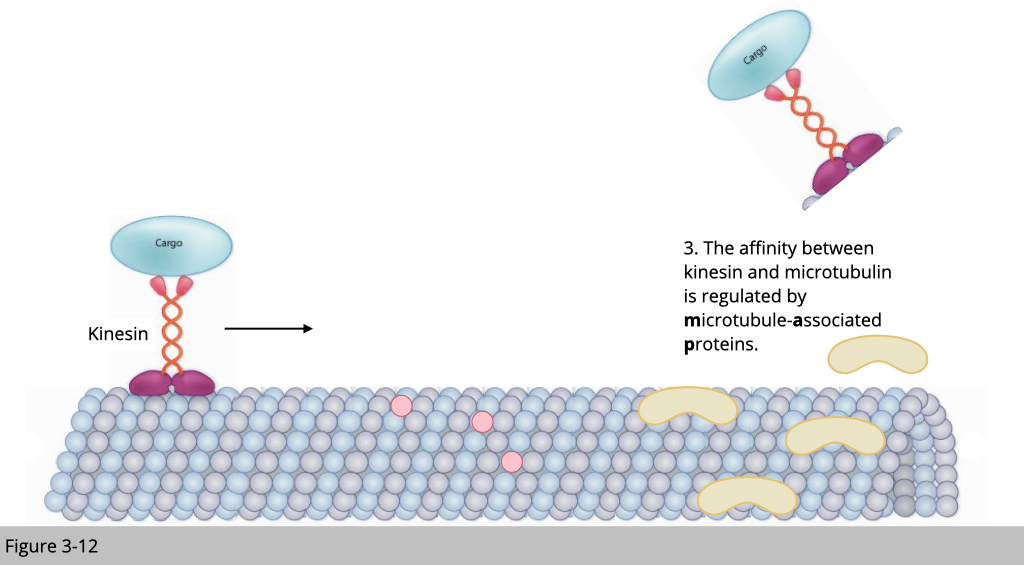
Within cells, motor and non-motor microtubule-associated proteins (MAPs) simultaneously converge on the microtubule. Motors must navigate a crowded microtubule that is decorated with non-motor MAPs. These MAPs have various roles in regulating microtubule dynamics, as well as in influencing motor transport. For example, the neuronal MAPs: MAP7 family members promote the binding of kinesin-1 to microtubules. Whereas, MAP2/Tau family inhibits kinesin-1 driven motility.
By utilizing different mechanisms such as post-translational modifications of motors or tubulin, and MAPs on microtubules to regulate the loading of motors on the microtubule, our cell could position the precise cargo delivery like what logistic companies did and do it even better.
There are two ways of transportation. First, the passive transport, substances simply move from an area of higher concentration to an area of lower concentration, which does not require the energy but it is slow. The second part of transport is active transport, which transportation is via motor protein using ATP as an energy resource.
Customized Shippment.

Similar to shipment in the real world, the cargo transportation within cells is customized.
- Whether the shipment is delivered outward or backward from the center of the cell to the proximate region closed to the plasma membrane.
- Should we take a ride on microtubulin for speed transportation or microfilament for local delivery?
- Should we hire only one driver- one motor protein at the time on the cargo for a specific delivery or should we hook all the motor proteins on the cargo for dynamic transportation or?
The direction of shipment and the location for the delivery decides which cytoskeleton to take and which motor proteins to be recruited for this shipment. Lastly, the most important information, what is the cargo? Depending on what the cargo is, cells will need to prepare the corresponding adaptor protein to load the cargo on its motor protein- the driver.
Express delivery.
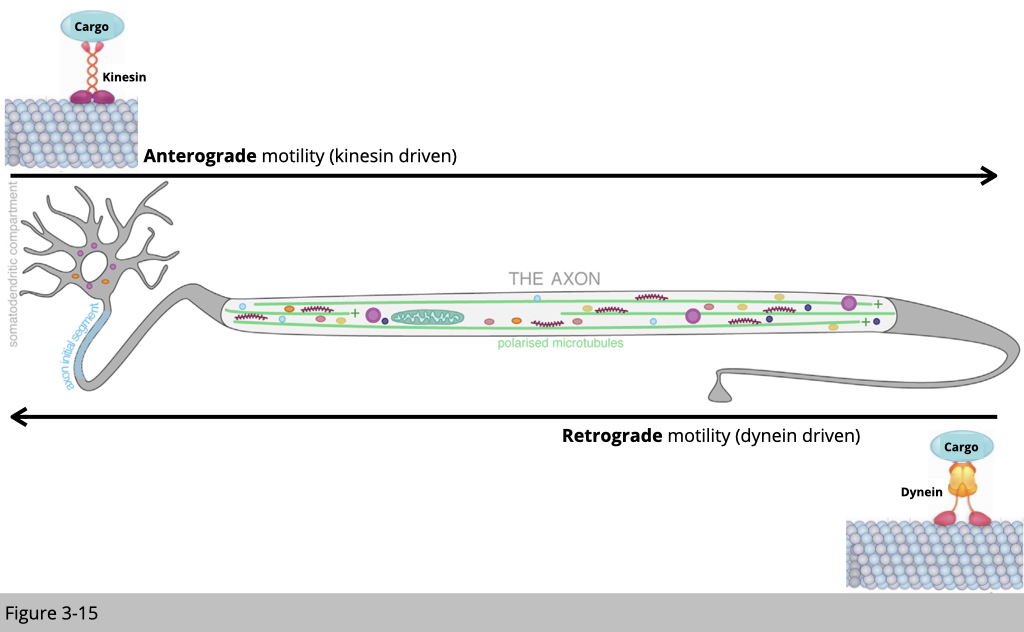
Using motor neurons as an example, there are two common active transportations. Microtubule-based transport often is used for long-distance transportation, such as from the cell body to the distal axon. Kinesin-1 carries the cargo out from the cell body. Dynein carries the cargo back to the cell body. Once the cargo reached the distal end of an axon, microfilament and myosin will help for the local delivery in the synapse and “secretion”, which the cargo can be diffused to the other cells. Vice versa, microfilament and myosin also help the cell to receive goody from outsides of the cell, so-called endocytosis.
Courier and shipping Services.
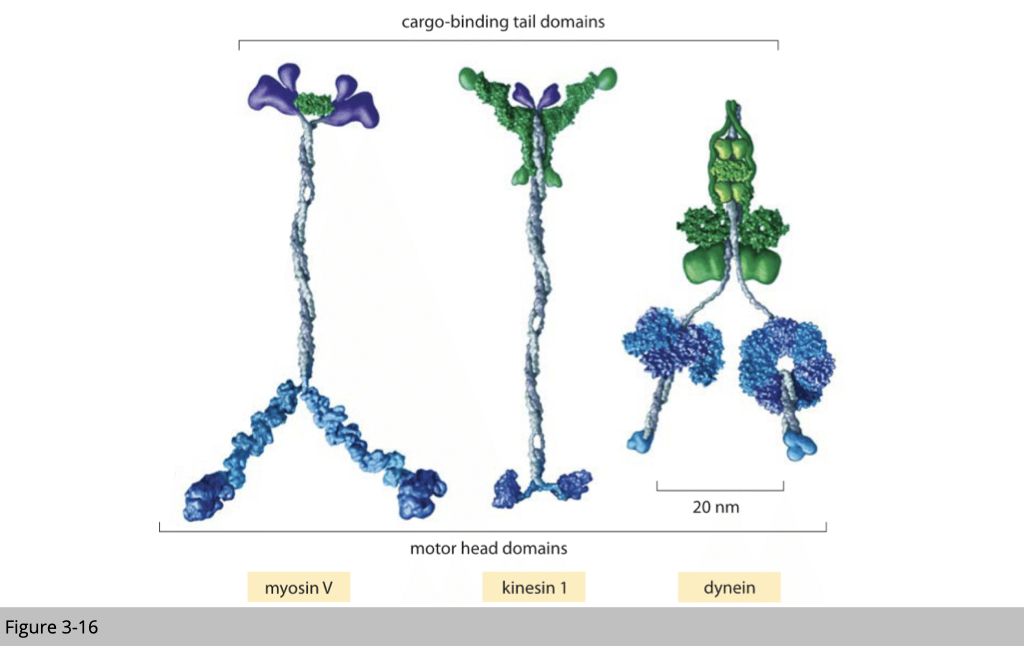
As reviewed, we have three types of motor proteins that can deliver the cargo. Myosin walk alone the microfilament, kinesin, and dynein walk on the microtubule.
The next question is how should we design a delivery? For those cargos that are transported bidirectionally such as mitochondria, does kinesin and dynein load on the mitochondria at different times? Or both kinesin and dynein are loaded on cargo at the same time, and they fight for each other to compete for the cargo carrier?
How could we study it?
We can purify the cargo and check whether the cargo is associated with kinesin and dynein together or separately.
Selective recruitment model.
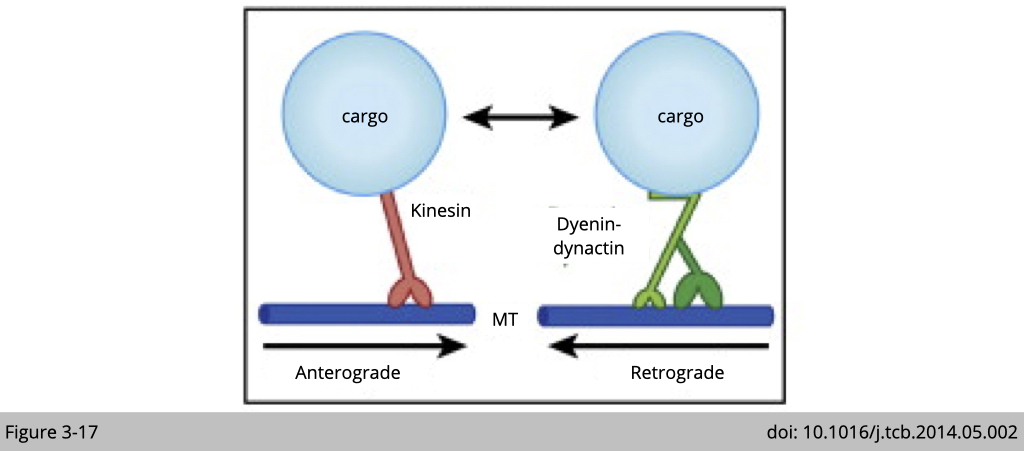
The simple model for the bidirectional transport is that cargo only binds to one type of motor at a time. When it binds to kinesin, the cargo transport from cell body to synapse. When it binds to dynein, it moves in the opposite direction.
Dynein and kinesin are copurified with endosomes in brain.
There are two classical ways to study the interaction between motor protein and cargo. One is differential and density gradient centrifugation, and the other is co-immunoprecipitation (co-IP), which we will discuss them in detail in the case of study I below.
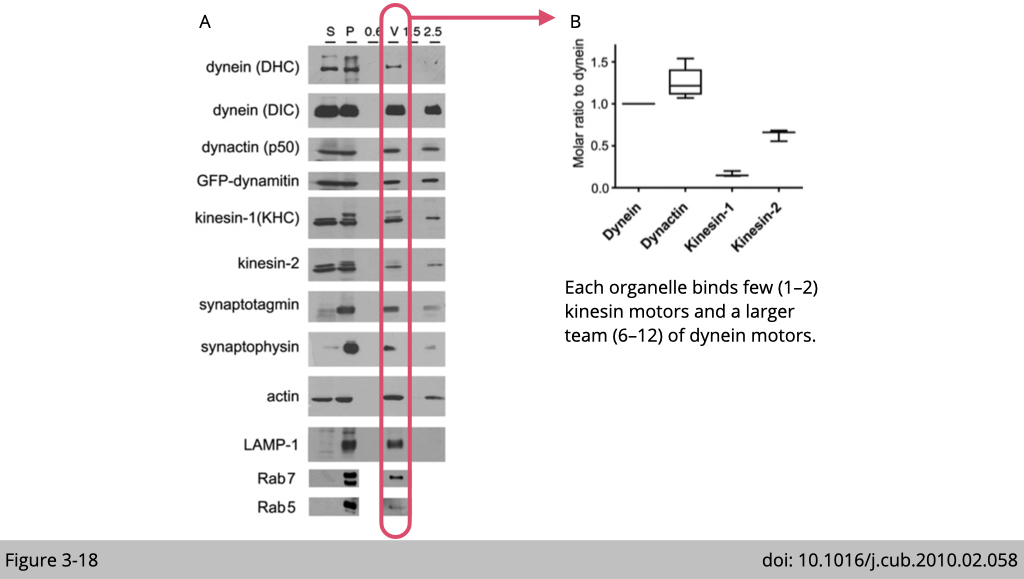
To investigate whether only one type of motor, either kinesin or dynein is bound to cargo at a time, we use the endosome transport in the brain as an example. Using a differential centrifuge and sucrose gradient, Erika L.F. Holzbaur’s group at the University of Pennsylvania isolated the endosome extracted from the mouse brain. The western blots of the protein lysates from initial cytosolic (S) and membrane (P), and the vesicle purification from a discontinuous sucrose gradient: the 0.6 M (0.6), 0.6/1.5 M (V), 1.5 M (1.5), and 2.5 M (2.5) were probed for different antibodies targeting motor proteins or markers for endosomes. (Figure 3-18A)
They found that the pure endosomes (markers: LAMP-1. Rab7 and Rab5) are highly associated with dynein and kinesin. By further calculating the ratio of the motor proteins presented in each fraction, they concluded that there is more dynein associated with the endosome than kinesin. Each endosome binds a few (1–2) kinesin motors and a larger team (6–12) of dynein motors (Figure 3-18B).
Tug-of-war vs Coordination models.
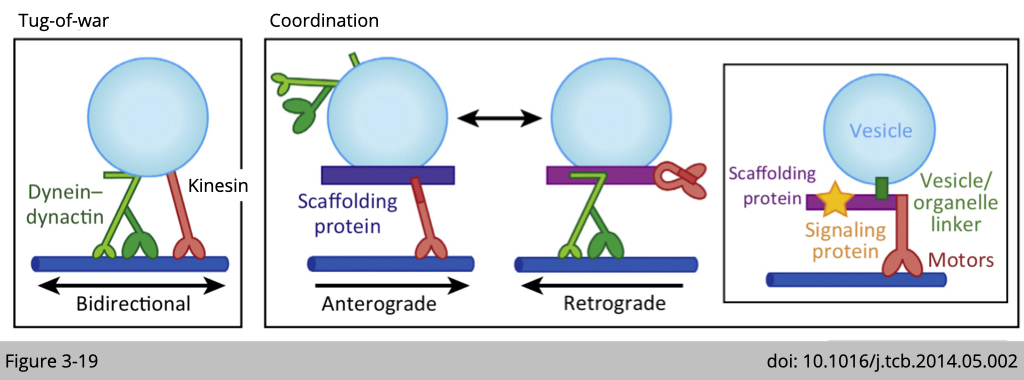
By the sedimentation assay showed above on Figure 3-18, we now know that both motors are loaded on the cargo at the same time. So how does it work? Does kinesin and dynein act like that twin brothers fight for moving all the time (Figure 3-19 Tug-of-war)? Or our cell use the adaptor system to regulate which motor will be active at certain time (Figure 3-19 Coordination)?
Huntingtin scaffolds kinesin and dynein motors in neurons.
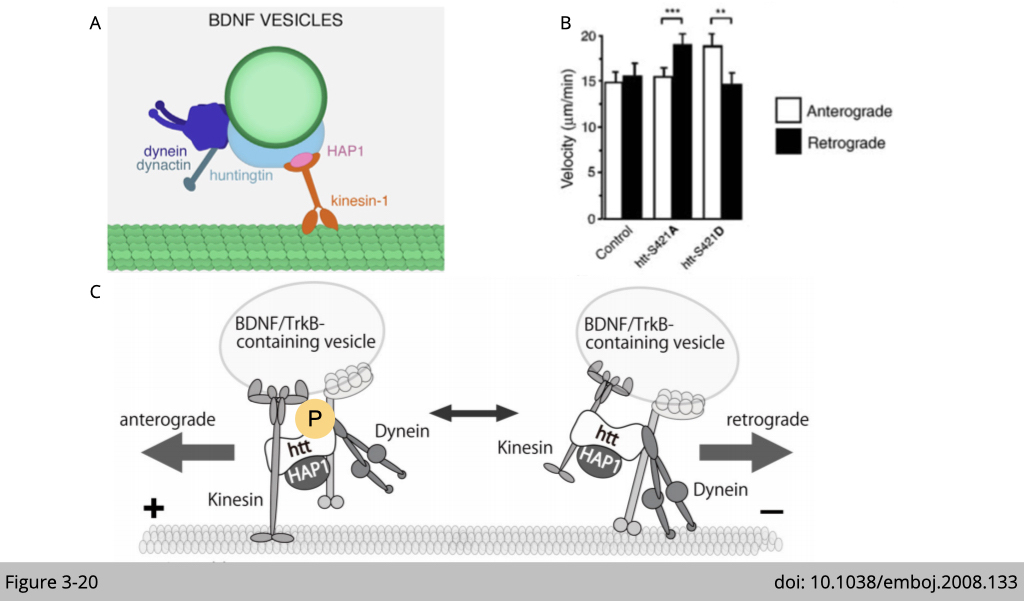
Using brain-derived neurotrophic factor (BDNF) as a marker of vesicular transport, Sandrine Humbert’s group at the Institut Curie in France, found that huntingtin (Figure 3-20A), the protein mutated in Huntington’s disease, is a regulatory factor for vesicular bidirectional transport in neurons.
This experiment was done in cultured cortical neurons and BDNF vesicles were labeled with a green or red fluorescent protein. Using end-binding protein 3 (EB3) coupled to GFP to trace the ‘plus’ end microtubule polymerization state, the movements of the EB3–GFP comets (90–95%) were directed distal to the cell body, indicating that outward movements correspond to ‘plus’ end-directed anterograde movements. Having determined the microtubule orientation in this experimental system, Humbert’s group tracked the BDNF vesicles in the cultured neurons and classified the intracellular movements as anterograde and retrograde transport according to the direction moved by the vesicles relative to the cell body.
They found that over-expression of huntingtin with alanine mutation mimics an unphosphorylatable S421, the BDNF vesicles moved faster in the retrograde direction than in the anterograde direction. In contrast, when the neurons were transfected with glutamic acid mutations of huntingtin mimics a constitutively S421-phosphorylated huntingtin, the vesicles moved significantly faster in the anterograde direction than in the retrograde direction (Figure 3-20B).
In summary, both kinesin and dynein binding to huntingtin and BDNF vesicles at the same time. When huntingtin phosphorylated active kinesin-dependent movement, the cargo is moving anterogradely. Vice versa, when huntingtin was dephosphorylated, the kinesin-dependent movement will be inhibited, therefore activate dynein-dependent retrograde transportation (Figure 3-20C).
What to ship via intracellular transport?
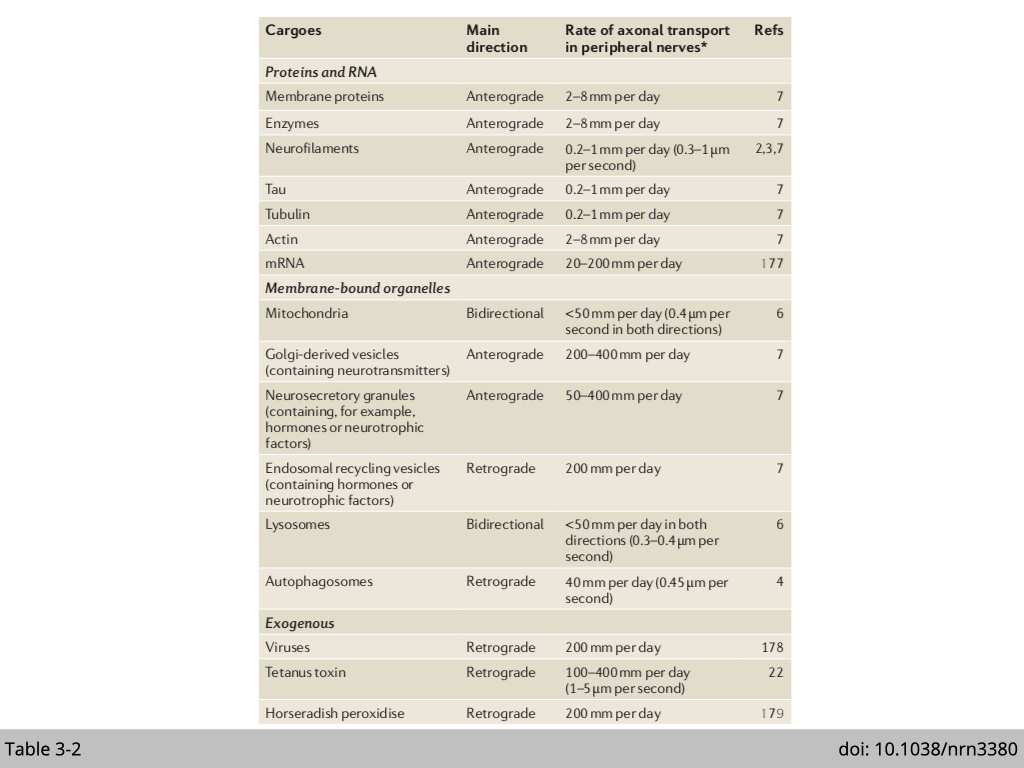
There is a range of goodies that can be shipped with cells, such as protein, mRNA, organelle, virus, and even nuclei (Table 3-2). Note that, anterograde transportation of mRNA and mitochondria is super important for neurons, especially motor neurons. A motor neuron is characterized by its long axon. It will be more efficient and productive to ship mRNA to the synapse and translate them to generate fresh proteins locally. Neurons mainly use mitochondria as energy resources. Moreover, for action potential to stimulate neuron excitement and neurotransmitter secretion both need a lot of ATP to mediate the process, neurons need a lot of mitochondria at the synapse for this energy demand.
Another example is nuclear movement in the skeletal muscle. A hallmark of the skeletal muscle cell is the position of nuclei at the periphery. Any newly fused myonuclei regenerated from skeletal muscle stem cell would need to migrate from a contral position within the myotube to the periphery for proper function. Mislocalized myonuclei have been associated with a variety of muscle diseases characterizing by reduced muscle size, muscle weakness, and decreased muscle function.

We have so many goodies to ship, and they all ship to different locations within cells. Do we have enough motor proteins delicate for specialized shipping? Or our cells adapt the tag system to mediate this specific delivery like FedEx?
In case study one, we will discuss two papers published by Thomas Schwarz’s group at Harvard Medical School, which studied axonal transport of mitochondria to synapses in fly as an example to demonstrate the role of “cargo-adaptor” in cellular transport.
Before reviewing the papers, I will brief you on two methods to help you with data interpretation.
Subcellular factions_ by differential centrifugation.
Shipment 101, the shipper must physically carry cargo for delivery. Meaning that motor proteins and cargo should be co-localized within crowded cellular environments. If our cells use an adaptor system to tag the cargo, then the “cargo-adaptor” should also have the same cellular localization with cargo and corresponding motor protein.
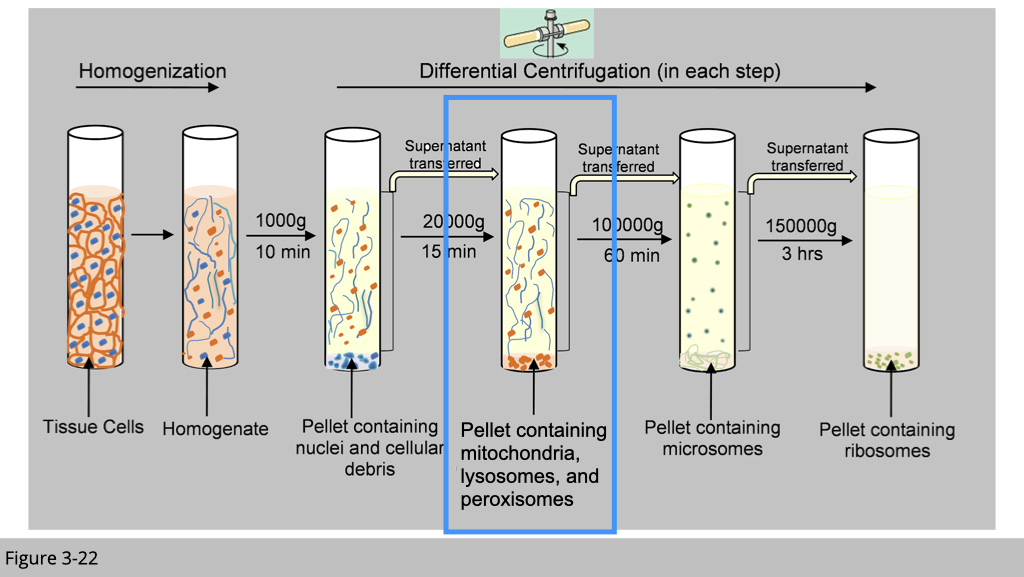
There are a couple of ways to detect the co-localization of proteins. The most common way is via immunofluorescence using antibody or fluorescent-tagged proteins to study protein subcellular localization, which we have introduced previously. The other “classical” experiment to exam the localization is through subcellular fractionation by using differential centrifugation as shown in Figure 3-22.
In general, the largest units experience the highest centrifugal force and move the most rapidly. At relatively low speed, large components such as nuclei sediment forming a pellet at the bottom of the centrifuge tube. Next at a slightly higher speed, a pellet of mitochondria is deposited. Finally, at even higher speed and with longer periods of centrifugation, first the small closed vesicles and then the ribosomes can be collected.
Subcellular factions_ by equilibrium density-gradient centrifugation.
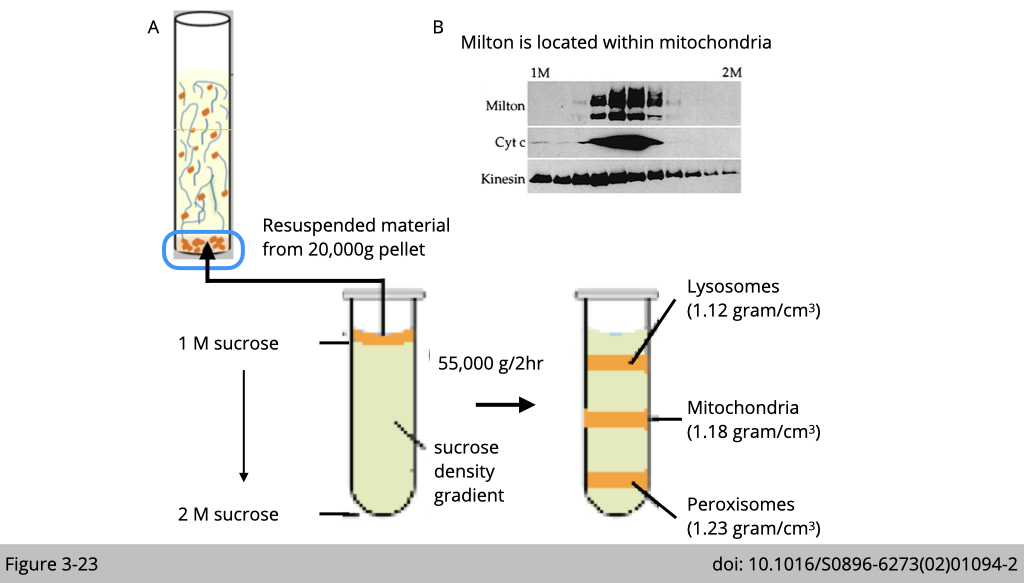
Yet, differential centrifugation does not yield pure organelle fractions. Vaious organelles not only differ in size but also in density. We can use equilibrium density-gradient centrifugation to further separate lysosomes, mitochondria, and peroxisomes, which separates cellular componenets accroding to their density.
In Figure 3-23A, the proteins from adult fly were extracted and postnuclear supernatant was loaded onto a 1-2 M sucrose gradient. The gradient was then spun at 55,000 g for 2 hrs at 4oC and each organelle will migrate to an equilibrium position where the density of the surrouding liquid is equal to the desnity of the indicated organelle and remains there. For example, the density of mitochondria is slight heaveir than lysosomes and lighter than peroxisomes. After running each fraction in western blot, we could find mitochondiral marker- cytochrome c only enriched in the middle fractions (Figure 3-23B).
Using equilibrium density-gradient centrifugation to finely separate cellular organelles, Schwarz’s group identified Milton co-sedimented with mitochondria complex- cytochrome C and kinesin. Whereas Kinesin heavy chain is more broadly distributed.
Protein interaction assays.
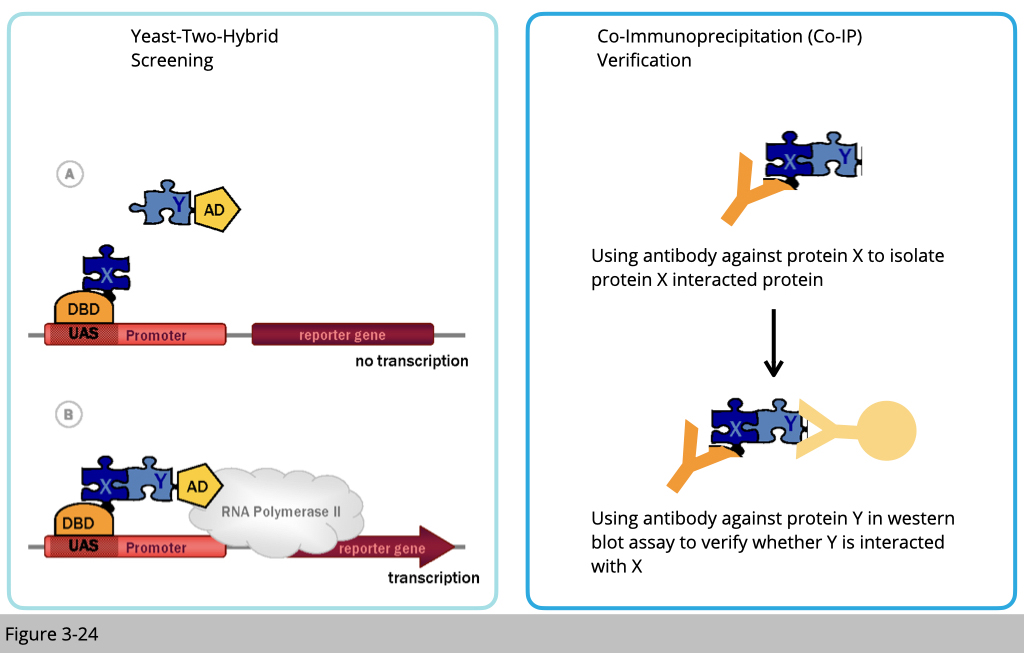
Next, they tested whether kinesin transport mitochondria through milton. If so, milton must directly bind to kinesin. There are many ways you can study protein interactions. Two of the classical ways (Figure 3-24): one is yeast two-hybrid.
Yeast two-hybrid is based on the reconstitution of a functional transcription factor (TF) when two proteins or polypeptides of interest interact. This takes place in genetically modified yeast strains, in which the transcription of a reporter gene leads to a specific phenotype, such as growth on a selective medium or change in the color of the yeast colonies. Two fusions (‘hybrids’) are constructed between each protein of interest and either the DNA Binding Domain (DBD) or the Activation Domain (AD) of the TF. The protein fused to the DBD is referred to as the ‘bait’ and the protein fused to the AD as the ‘prey’. Upon interaction between the bait and the prey, the DBD and AD are brought in close proximity and a functional TF is reconstituted upstream of the reporter gene.
The other way to detect protein interaction is through co-immunoprecipitation (Co-IP). Using an antibody to pull down the complex of protein x, if x-y proteins have interacted, then we shall detect protein Y presence in the complex of protein x.
The design of immunoprecipitation- Specificity of antibody bonding
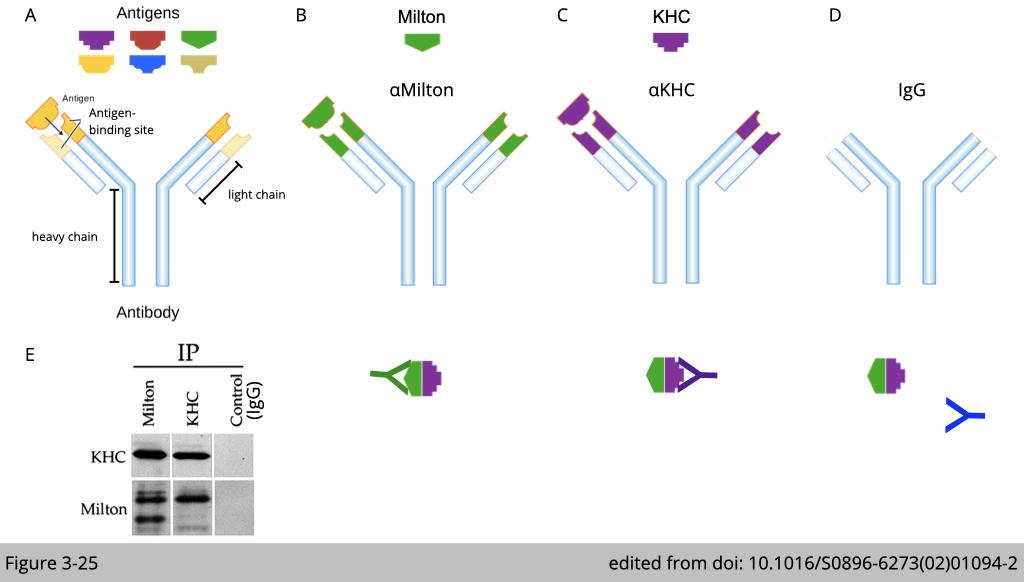
Figure 3-25 used Co-IP to confirm the endogenous interation between milton and kinesin in cell. If milton interacts with kinesin, we pull down the complex of milton, we shall detect kinesin in the complex of milton (Figure 3-25, B and E). Vice versa, we can pull down the complex of kinesin, we shall also detect milton in the complex of kinesin (Figure 3-25, C and E).
Note that, this experiment required an important control pull-down analysis, that we need to show the antibody pull-down is specific. The control pull-down of antibody backbone- IgG should not detect kinesin or milton in the Co-IP lysate (Figure 3-25, D and E).
Milton interacts with kinesin: mapping the kinesin-binding regions of milton
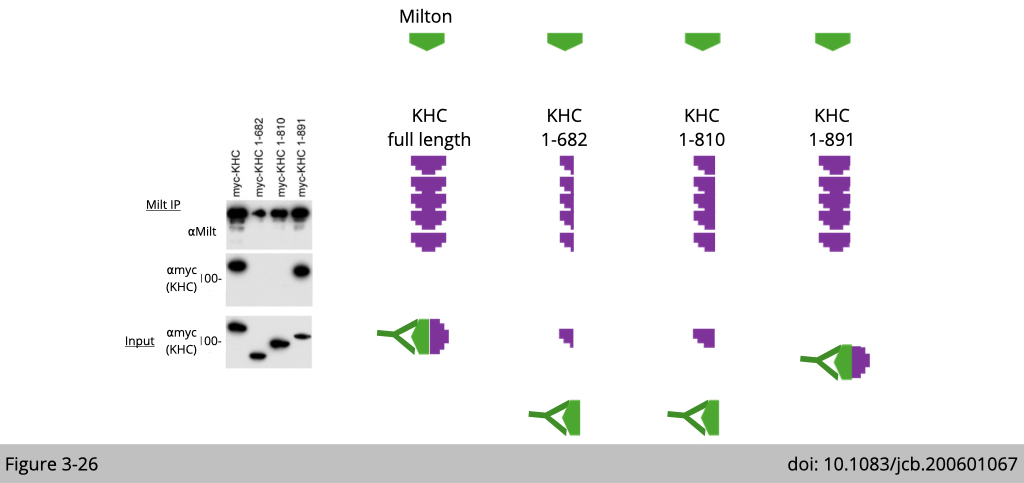
Now we know milton not only co-localizes but also interacts with kinesin. Next, we need to know which part of kinesin interacts with milton, which will suggest how milton mediated the specificity of mitochondria transport via kinesin.
This experiment was designed as such: the full-length milton was cotransfected with each of three truncated myc-tagged kinesin heavy chain (KHC) constructs and then pull down with antibody of milton. In Figure 3-26, you can see that milton is associated with both full-length KHC and KHC lacking the last 64 amino acids of the tail domain (mycKHC 1–891), but not with KHC lacking the entire tail domain (mycKHC 1-810) or a larger deletion (mycKHC 1-682). Therefore, the KHC tail region 810–891, but not 892–955, was necessary for associating with milton.
Note that this experiment was performed by over-expression of different KHC constructs. We need to make sure the over-expression of these construct have a similar level (Input) for proper interpretation of Co-IP result. For example, the expression of mycKHC 1-682 and mycKHC 1-810 are a little bit higher than mycKHC 1-891, yet only mycKHC 1-891 can be detected in the complex of milton (Milt IP). Therefore, we are certain that the absence of mycKHC 1-682 and mycKHC 1-810 in the complex of milton is not due to the lower expression.
Milton interacts with kinesin: mapping the milton-binding regions of kinesin
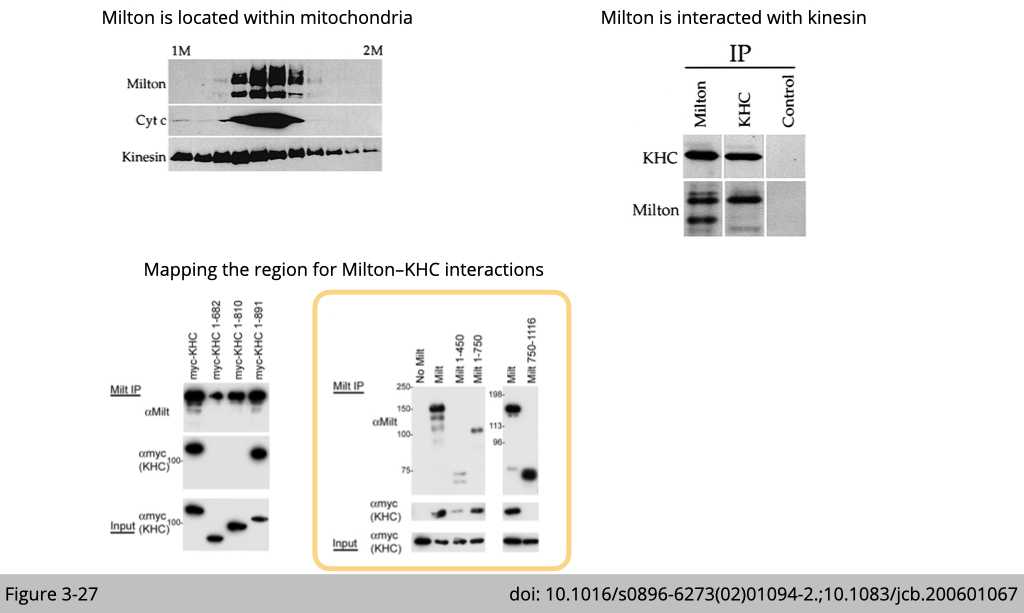
So now we know kinesin is mainly associated with milton using its tail domain (810–891), where they used to carry the cargo. Next, we need to map which part of milton interacts with kinesin.
Milton comprises 1,116 amino acids with no recognizable structural motifs. The pull-down experiment (Figure 3-27) shows that milton 1–450 was sufficient to associate with myc-KHC. Milton 1–750 also coimmunoprecipitated with KHC, but the COOH-terminal domain of milton (Flag-tagged milton 750–1,116) did not.
We have information on the localization and interaction of Milton and kinesin. The final question is what this interaction relevant to physiology?
Axonal transport of mitochondria requires milton to recruit kinesin.
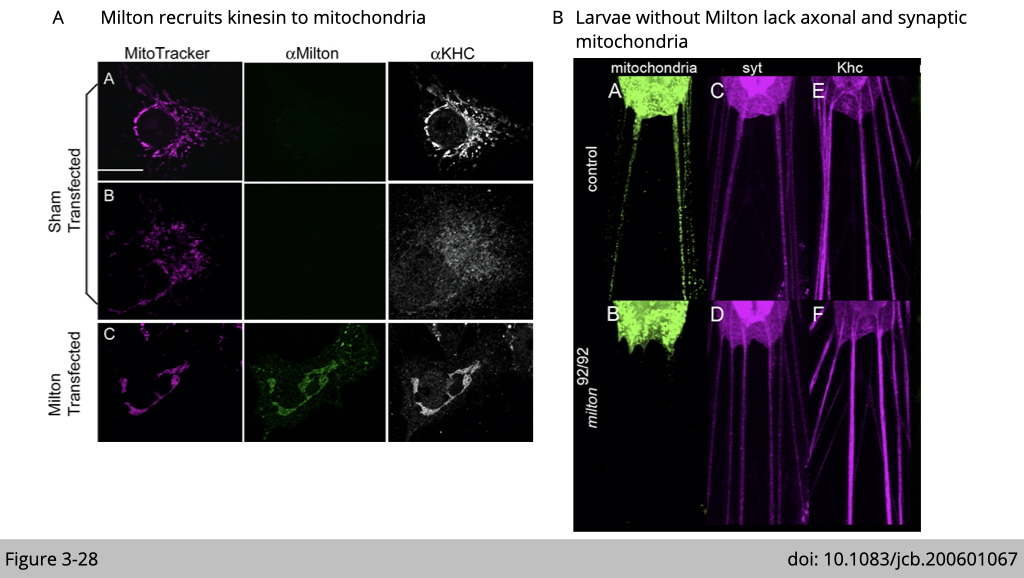
Figure 3-28A showed that mammalian cell line either transfected with fly milton or not. The endogenous KHC was typically cytoplasmic distribution in untransfected cells, although KHC was also observed on mitochondria. Upon transfection with fly milton, firstly, we can observe that the signal of milton was located exclusively on mitochondria. Second, upon transfection with milton, the KHC became highly enriched on the mitochondria, with little remaining detectable elsewhere in the cell. The interpretation of this immunostaining result is that milton recruits KHC to relocate on mitochondria.
Whether milton is really the adaptor for kinesin to carry mitochondria in the axon? Schwarz’s group examined the mitochondria distrution in larva (Figure 3-28B). The larva is the main feeding stage of the fly. Mitochondria were fused with GFP for visualization in a neuron. In control larvae, numerous mitochondria were present in the axons. However, in milton mutation larvae, axonal mitochondria were absent. This defect is selective for mitochondria transportation since the continued presence of signals for kinesin and the synaptic vesicle marker synaptotagmin in axons were not affected.
Cargo adaptor.
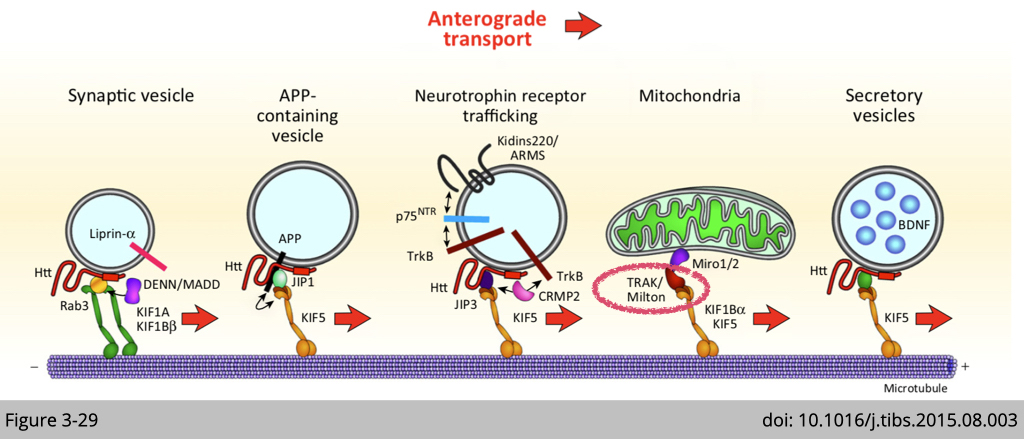
These two elegant piece of work makes milton on this cute cartoon in Figure 3-29. In summary, our cells did adapt the tag system to mediate the specificity of cargo delivery. Many of them are waiting for you to discover!